
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
This report has been reproduced directly from the best available copy.
Available to DOE and DOE Contractors from the Office of Scientific and Technical Information, P. O. Box 62 Oak Ridge, TN 37831; prices available from (423) 576-8401.
Available to the public from the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Road, Springfield, VA 22161.
An experimental investigation of the relationship between tritium surface activity measurements and the accompanying tritium concentration through the bulk of concrete exposed to tritium at two locations at the DOE Savannah River Site has been completed. The goal of this work was to increase the understanding of i) the relationship of measured surface activity to the underlying bulk tritium concentration in concrete, and ii) the nature of tritium contamination in concrete generally. Such an understanding could lead to faster, less expensive characterization for waste disposal in deactivation and decommissioning projects. Specifically, the following tasks were accomplished.
1) Two new tritium surface activity monitoring systems, the Surface Activity Monitor (SAM), a real-time tritium activity monitor, and the (E-Perm) electret passive integrating detector, were found to be effective surface tritium activity monitors for concrete. The electret could detect tritium to significantly lower levels than the SAM, however at sufficient tritium surface activity both systems proved to be effective. In addition, a portable liquid scintillation counting device (Lumi-Scint) was successfully evaluated as a field deployable tritium assay for surface smears.
2) Simple impact drilling of concrete, with the aid of a hammer drill to collect pulverized concrete in stages during drilling, was a useful and inexpensive technique to remove samples through the concrete bulk. Impact drilling allows through-thickness measurement of tritium concentration in depth increments as small as 1 inch. Traditional core drilling is much slower and more expensive, and generates much more secondary radiological waste.
3) A new nitric acid leaching technique was employed to prepare concrete samples for tritium assay by liquid scintillation counting. These samples were also used to characterize the silicon and calcium content with depth, employing inductively coupled plasma emission spectroscopy (ICP-ES).
4) The concentration of tritium in concrete at both locations, one a heavy water processing facility (420-D) and the other a former tritium facility process area (234-H), was found to be nearly constant with depth. Both locations revealed slightly lower concentrations at the surface, more so at the 420-D location. The 420-D location was exposed to the outside weather, suggesting that weathering reduced the tritium surface concentration, probably by diffusion and isotope exchange with rain water and water vapor in the air. Slight variations in the concentration with depth in the 420-D location can be explained by the presence of low-calcium aggregate in the concrete: by normalizing the tritium activity to the calcium concentration at each depth, the variation was significantly less. The concrete assayed in 234-H was a grout, containing no aggregate, and had a uniform tritium concentration both before and after normalization to the calcium concentration.
5) A literature survey of tritium in concrete reveals several studies in which concrete is exposed to tritiated water vapor or tritium gas. The concentration of tritium in these studies decreases rapidly with depth from the exposed surface inward. This is significantly different from the experimental findings in both locations during this study. The relatively uniform concentration of tritium found in the bulk in this study is believed to occur because the tritium was in the liquid state: either tritiated heavy water at 420-D or either tritiated water or tritiated oil at 234-H. (The history of spills at the 234-H location is uncertain, so details of the liquid or liquids causing the contamination is unknown.) The constant concentration of tritium results from the comparatively rapid transport of liquid into the surface capillary pores of concrete, which is well substantiated in the literature, combined with the relatively slower isotopic exchange of tritium in the capillary pores with hydrogen in the gel pores and in water of hydration in the cement gel itself. A uniform and stable concentration profile results from subsequent diffusion, with the exception that weathering can reduce the surface concentration over time (discussed above).
6) A model was created relating the surface activity as measured by the presence of beta-particle induced ionization, which is the detection mechanism used by both the SAM and the electret, with the bulk concentration. Specifically, this model postulates that the detection of tritium decay ionization is related to the high effective surface area of concrete at the surface (stemming from the pores in concrete), such that the surface activity measured by the SAM or electret can be directly related to the bulk concentration near the surface through estimates of the areal density at the surface.
The various forms of cement and concrete are the most common industrial building materials. They are particularly useful in the nuclear industry because they are strong, durable, inexpensive, and shield radiation well. However, cement has some unwelcome properties for some nuclear applications. These arise from the porous nature of prepared cements and the ever-present water in them. This can often result in the incorporation and retention of some radioactive species, especially tritiated water, that could be subsequently released into the environment. While many nuclear facilities may be contaminated with tritium, those in the Department of Energy (DOE) facilities associated with the production of tritium for weapons programs, or with Canadian Deuterium Uranium (CANDU) reactors are of principal concern because of their high levels of potential tritium contamination.
These facilities will either eventually have to be dismantled or already have been. In both cases, proper characterization of the amount of tritium retained in the concrete is required for waste disposal. Detecting tritium is problematic in most materials, because of its weak beta energy and absence of gamma rays, and so can be very expensive. The hydrophilic nature of cements and concretes makes them among the most difficult to properly characterize. Tritium contamination on their surfaces is usually easy to detect from analysis of smears (wipes), but only with a high degree of variability and uncertainty in relating the smear count to the actual surface activity. To detect tritium in the bulk, most often cores are drilled for subsequent laboratory analysis. Core drilling requires expensive special equipment, often spreads contamination, presents handling and storage problems, and is slow. Coring equipment usually requires a cutting or cooling fluid. This can spread contamination and generate secondary waste. Cores are also expensive, particularly on a large scale; hence they usually must wait until formal deactivation and decommissioning (D&D) activities are begun. With such delay, essential facility operators and experts may no longer be available, and their knowledge of incidents and precise locations and circumstances needed to best do the characterization job is lost.
Despite the effort required by the "traditional" core drilling and surface smearing characterization methods, their success in proper tritium characterization is not a certainty. At the Savannah River Site (SRS), missed contamination in structural concrete at a retired tritium facility resulted in lengthy delay and significant added expense to its D&D project. The difficulty of characterizing tritium in concrete results partly from the complex and poorly understood behavior of tritium in cement and concrete. With sufficient sampling, detailed knowledge of these complex properties is unnecessary for tritium characterization. (Tritium in cores cannot stay hidden when subjected to proper preparation and analysis.) Avoiding such problems in future site operations and D&D projects and reducing the cost of tritium characterization are the prime motivations behind this work. With better and faster new tools and improved understanding of tritium in concrete, a much cheaper and more useful characterization may be possible at the time of facility deactivation.
There is a considerable body of research literature dealing with the methods and measurements of tritium in a variety of materials including cement and concrete. Dickson [1] has summarized much of this reported prior to 1990, and relevant work continues to be published. New surface tritium activity measurement devices that are currently being developed, together with more simplified methods for collecting and analyzing bulk concrete samples, offer the promise of cheaper, better and faster tritium characterization. This paper describes a study to demonstrate these new methods at two different SRS tritium facilities: one at a tritiated heavy water facility (420-D), the other at a tritium facility (234-H). With these techniques to improve the traditional ones now used in D&D, empirical data, so far, suggest that measured tritium surface activity can be used to infer that in the underlying bulk with accuracy sufficient for most characterization purposes. Demonstrating the hypothesis that surface activity, measured by ion chamber type detectors, varies proportionally with the level of bulk activity is a primary purpose of the work described in this paper.
Dickson [1] reviews the literature of tritium interactions with a wide variety of building materials. Numata et al. [2] performed thermogravimetric analysis by heating cement specimens of different curing times, and monitoring weight changes and offgassing of the cement. They found that the water bound in cement could be described as being distributed in four different locations: pore and capillary water (67±7% of total water) , which desorbed below 200°C; water of crystallization (20±3%), which desorbed between 200-450°C; and the remainder was split into two types of constituent water in calcium hydroxide and calcium silicate hydrate, which desorbed between 450-550°C and 550-850°C, respectively. When specimens, previously wet-cured, were exposed to HTO vapor for up to 120 days, a steady state ratio between these waters was reached after about 50 days. About 80% of the HTO remained in the pore water even after 100 days of exposure, suggesting that tritium exchange between free and bound water is fairly slow.
Another Numata et al. paper [3] also demonstrated that tritium vapor and liquid exposure of various cement formulations could be fitted and interpreted by standard diffusion and permeation equations. This work was on well-characterized small samples, and no experiments on concrete were performed. In another report [4], Numata translated and interpreted the earlier work by Saito [5] on contaminated concrete in a CANDU reactor. Results were similar to the controlled cement work. Noteworthy in Saito's work is that small samples of drill powder were analyzed instead of massive concrete cores. The powder samples were leached in water for a week and analyzed by liquid scintillation. Numata states [4] that there was negligible loss of tritium from the powder samples in this study [5].
Another quantitative study on actual structural concrete exposed to tritiated water (HTO or DTO) vapor was reported by Krasznai [6] in connection with the decommissioning of a CANDU reactor. Here through-wall concrete cores were taken in sections 20-30 cm long and profiled for tritium. The tritium profiles in both regular- and high-density concrete showed a rapid decrease from the exposed surface inward. This behavior was seen in all the studies above and seems to be indicative of vapor diffusion into the solid concrete.
The importance of the complex cement hydration properties relative to the effects on tritium leaching behavior was pointed out by Matsuzuru [7]. The authors noted that water that is not chemically bound as hydrated water is physically retained as capillary and gel water in hardened cement. This gel and capillary water in the pore structure of the cement is free to exchange with external water which may quickly penetrate into the capillaries and then slowly diffuse into the gel pores of the matrix.
The chemistry involved in the hydration of cement and its subsequent leaching is further clarified in Zamorani [8] and Eichholz, et al. [9]. While these papers are specifically concerned with the leaching of various radioactive species including tritium from buried waste in a cement matrix due to water permeation, the chemistry and migration mechanisms discussed are useful in understanding the behavior of tritium in cement materials in general. Zamorani [8] describes the leaching process, primarily for Ca2+ ions, from the calcium silicates in hardened cement by excess water. Initial hydration of cement produces Ca(OH)2 and calcium silicate hydrates, which form a gel. The gel is maintained as long as Ca(OH)2 is present, and the gel acts as barrier to slow the influx of water and outflow of Ca(OH)2 around the solid central clinker (constituent particle of cement). Although Ca(OH)2 is slightly soluble in water, it cannot be removed from the solid bulk without a flow of water through the cement pores. When the pores of the cement are saturated with static water, the solubility product of the hydroxide is reached stopping further dissolution and a equilibrium is maintained. Under such an equilibrium, hydrogen exchange between the pore water and gel is not stopped, but merely slowed by diffusion of tritium and by the limited solubilities of the cement hydration products involved. As a result, tritium initially present only in the pore or gel water will exchange with the other solid-phase species, albeit relatively slowly.
These effects and the possible mechanisms for the mobility of tritium in concrete are broadly explained by Eichholz, et al. [9]. This laboratory study was started by briefly exposing dried concrete cylinders to a top layer of HTO, with the intent to measure the time for the pulse to reach the other side. This proved to be impossible because the pulse was immediately absorbed and retained at the top of the cylinder. Further study of absorption of standing water by dry concrete confirmed that a 3.75-cm tall cylinder reached 85% saturation in just over 10 hours of exposure, and a 15.5-cm one took about 130 hours. They conclude that in the absence of any surface coating, dry-cured cement will absorb much standing water and retain it over long periods, in the filled pores.
Two recent papers tend to explain this rapid sorption of water. Martys and Ferraris [10] looked at the rates of water absorption of oven and bench-dried mortar and concrete specimens. They found that although oven-dried samples absorbed nearly three times the water of air-dried ones, both were characterized by a very rapid initial uptake followed by a much slower one later on. They suggest the observed sorption is caused by pores of two different size scales. The larger ones exist mostly at the air-surface interface and contribute to a rapid capillary suction. These are particularly susceptible to enlargement by leaching and drying from weathering effects. Much smaller pores exist within the bulk material and are responsible for the much slower sorption by diffusion. This difference in the water sorption between the near surface and deeper bulk material was confirmed by Beyea et. al. [11] using magnetic resonance imaging. This technique was able to show the depth profile of evaporable water with sub-millimeter resolution. Profiles of prepared concrete samples with varying water-to-cement ratios and curing times all showed large amounts of capillary flow at early air-drying times and diffusive drying fronts which dominated at longer times. Most of the capillary action occurred in the first few millimeters from the surface after one-day drying with significant water loss down to twenty millimeters for longer times (7 and 90 days of air-drying) for samples with increasing porosity. Presumably, resorption of liquid water would be largely the reverse of desorption seen during evaporative air-drying.
Most of the literature cited thus far deals with experiments designed to explain the uptake or release of tritium from contaminated cement paste or concrete from a diffusion view point. While these are useful for material comparisons and estimating tritium gain or loss under certain conditions, they address little of the chemical mechanisms involved. Much insight to the chemical state of concrete is discussed in Lea [12]. Neville [13] has more information relevant to states of water and mechanisms of its migration in concrete. Neville's discussion on the structure of hydrated cement is excellent [13]:
"Fresh cement paste is a plastic network of particles of cement and water, but once the paste has set its apparent gross volume remains approximately constant. At any stage of hydration the hardened paste consists of hydrates of the various compounds, referred to collectively as gel, of crystals of Ca(OH)2, some minor components, unhydrated cement, and residue of the water-filled spaces in the fresh paste. These voids are called capillary pores, but within the gel itself there exist interstitial voids, called gel pores."
"Since most of the products of hydration are colloidal, during hydration the surface area of the solid phase increases enormously, and a large amount of free water becomes adsorbed on this surface. If no water movement to or from the cement paste is permitted the reactions of hydration use up the water until too little is left to saturate the solid surfaces, and the relative humidity within the paste decreases. This is known as self-desiccation. Since gel can form only in water-filled space, self-desiccation leads to a lower hydration compared with moist-cured paste."
From the above discussion, it is now possible to hypothesize about the behavior of tritium in cements and concrete. Most cements never fully hydrate unless more water is introduced after setting. Self-desiccation dries the capillaries, and further hydration virtually stops, leaving some dry cement that will react only if more water enters. Neville shows that at typical water/cement ratios of about 0.5, as much as 25-30% of the cement remains unhydrated unless more water is absorbed. As a result, most hardened cements can take up condensed water contacting the exterior of the concrete very rapidly. If enough additional water is absorbed, however, hydration products start to fill the capillaries and they become segmented rather than continuous. This condition effectively ends the hydration process.
In general case, both tritiated water and elemental tritium can migrate in cement and concrete. Five potential pathways for the migration of tritium in cement are [9]:
If the absorbed condensed water from the environment contacting the concrete contains HTO or T2O, tritium is integrated into the new gel that forms at unhydrated sites in the old gel mass. The gel containing tritium is otherwise indistinguishable from that with protium. Neville further points out that below relative humidity of 45%, the capillaries are emptied of water. Under the same conditions, the gel pores are not emptied of water. The gel pores are only 15-20 Å across and water primarily adsorbs onto the surface of the gel. Surface forces in the small gel pores bind water with significantly more energy than in capillaries. Of the various types of water in hardened cement, only water in the capillaries is relatively free to move. But even this freedom depends on prevailing environmental conditions. In the presence of high humidity, capillary water will be stagnant unless there is hydraulic flow (flow due to external water pressure difference across the concrete structure), since the capillary pores are filled with water. In low humidity, capillary water is lost leaving the bulk much as a dry sponge, able to soak up water given the opportunity. The other non-capillary waters may be thought of as being fixed to varying degrees. Tritiated water introduced into the capillaries will, especially with the help of liquid water, percolate and exchange with other hydrogen species throughout the capillaries and eventually reach a fairly uniform concentration throughout the bulk.
The case of an airborne exposure of concrete to tritium gas or tritiated water vapor is quite different from the exposure of concrete to condensed water discussed above. The amount of tritium contained in air, aside from extraordinary conditions, will be nearly a thousand times lower than that in a liquid from density considerations alone. Tritium concentration profiles in cement or concrete exposed in this manor invariably show a rapid decrease in concentration with distance from the exposed surface (discussed above [2, 3, 4, 5]). This type of profile is typical of diffusion controlled processes. Unhydrated cement can readily react with the oxide form as the tritiated vapor will diffuse very rapidly into the capillary pores of the cement. However for elemental forms, a different mechanism likely exists. Exchange with normal hydrogen in the water of the gel phase, aided by surface forces to help break bonds might be an explanation. But unmentioned in any of the reviewed literature are radiolysis and radiation effects. Although the beta decay energy released during tritium decay is very low (18 keV maximum, 5.7 keV average), it is still about 1000 times that needed to break chemical bonds. Broken bonds caused by radiation acting on moisture in air is a primary mechanism for the conversion of HT and T2 into HTO. This effect should be even more important in cement, because of the close proximity of matter inside the small capillaries and pores of hardened cement. In addition, the species T2 produces a reactive tritium atom or ion when the other tritium nucleus decays. These are all highly reactive species, which will quickly react with water and the other cement constituents.
These five tritium migration pathways listed above involve both redistribution of tritium within the bulk material and releases to the surroundings. Both diffusion and mass flow of tritium are involved, and once at the surface boundary, physical and atmospheric conditions may further affect surface activity. Understanding all of these mechanisms from an atomistic viewpoint is beyond the scope of this work. Instead, we focus on the qualitative understanding outlined above, which leads to an empirical relationship relating the surface and bulk tritium activity. This relationship may allow surface activity measurements to be reliable estimators of bulk tritium burden, adequate for waste disposal characterization.
Probe surveys are used as a primary method of contamination detection of many isotopes in nuclear facilities, but not with tritium. The low energy of tritium betas requires a windowless detector be used to measure tritium surface activity. Such devices are available but they often encounter operational problems with in-situ measurements. Equipment size, need for counting gas, and effects of changes in ambient conditions, which invariably will occur, have all limited the usefulness of past instrumental approaches. Instead, the main method of detecting tritium contamination has been by smears of the surface in question followed by liquid scintillation counting (LSC) of the smear. The detection of tritium by LSC has been significantly improved, and, presently, is still the most sensitive method available. Unfortunately, the amount of activity collected on the smear is a highly variable and inexact process, which is influenced by many physical and environmental factors as well as the person doing the smear. Also, the best LSCs for tritium are not portable and require a permanent laboratory facility.
Several new technologies have emerged in the last several years with the potential for more reliable in-situ measurement of tritium surface contamination. One is called an electret [14]. This is a hockey-puck size electrostatically-charged disk mounted in a conducting plastic housing which has a opening to admit radiation or the ion current that radiation produces. When placed on a contaminated surface, the electret acts as a passive ionization chamber. Its sensitivity depends on the length of time available for the measurement; longer exposures result in lower detection limits. By measuring the change in electrostatic voltage between deployment and recovery (using a separate auxiliary reader), and the time of exposure, a prior factory calibration is used to determine the activity of tritium on the tested surface. Tritium activities down to about 0.1 nCi/cm2 can be measured by long exposures (48 hours). For much higher surface activities, measurements of a ten minutes to several hours minutes are generally adequate. A high range electret, which is ten times less sensitive, is also available for more highly contaminated surfaces.
Another detector measuring the ion current due to tritium decay is the Surface
Activity Monitor (SAM) [15]. The SAM tested in this study is about the size of
a flashlight and weighs about 600 g. Its housing contains a charged collection
plate and a sensitive electrometer. When placed open-side down on a
contaminated surface, it can reliably read tritium activity of about 10
nCi/cm2 and greater, directly, within a few seconds. This is much
less sensitive than the electret, but it is also much faster. Both the SAM and
electret have open circular measurement windows of about 10 cm2.
A portable LSC, called the Lumi-Scint [16] is available, which is small enough
for field use. It is about the size of a shoe-box and weighs about 5 kg. The
instrument uses a single photomultiplier tube (PMT) to measure light from a
sample-containing vial introduced into its sample chamber. Because there is
only one PMT (rather than the normal two), photoluminescence from exposure of
the cocktail to direct sun or fluorescence lighting is a problem. With care
this can be largely avoided, so a filter paper smear placed into a vial of most
cocktails can be measured down to about a few thousand dpm of tritium with a
minute or two of counting. Since smears are normally taken over
100-cm2 areas, this method has the best sensitivity among these
three that we tested, depending on the assumed sampling efficiency of the smear.
Locations at two different SRS processing facilities were selected for this study. The original intent was to select one likely to have been contaminated by exposure to aqueous tritium and the other to elemental tritium. On the basis of laboratory LSC analyses of survey smears taken at likely places in the two facilities, one area outside the 420-D Building (heavy water processing) and another in the 234-H Building (former tritium gas processing) were selected. The 420-D area was in the concrete sump beneath an outdoor storage tank for DTO-contaminated high purity D2O reactor moderator. This tank had a history of leaks over many years. Present surface contamination at this location is well below 10,000 dpm/100 cm2, the threshold activity for a Contamination-Area classification at SRS. The 234-H area is a section of grouted floor, immediately adjacent to a process cabinet, which was contaminated to greater than ten million dpm/100 cm2. This area in the building processed elemental tritium, and is no longer active.
A 15-cm diameter work circle was marked at each of the two study areas. The work circle was stripped of any visible surface sealant material and buffed with a disk grinder to yield a relatively flat, smooth surface in preparation for the surface measurements. This grinding was also necessary to remove the surface layer of CaCO3 that can form due to the reaction of atmospheric CO2 with the Ca(OH)2 formed in hydrated cement. Loose dust or debris from the grinding was removed by vacuuming.
A series of comparative surface measurements were made using the detectors described above. Different parts of the work circle were first measured with the SAM to determine the uniformity and level of surface contamination. The SAM detected surface contamination for the relatively high activity level at 234-H, but the SAM's sensitivity was inadequate for the low level at 420-D. Next, electret measurements and Lumi-Scint LSC measurements of smears were made in both work circle locations. At 234-H, a high-range electret was placed in the center of the circle and left overnight. Following this, two smears (Whatman glass-fiber filter paper) were made by successively wiping the entire area of the circle and placing each in labeled, pre-filled LSC vials. The Lumi-Scint was used to count the two vials once just after they were taken and then again two hours later. During this time, a low-range electret was placed in the center of the circle for a second measurement. There was an activity increase of about 20% over the 24-hour span between the two electret measurements. No further increase occurred after a second 24-hour wait. This presumably reflects reestablishment of equilibrium after the surface was disturbed by the prior grinding and vacuuming. In general, this implies that about a day or two should pass between surface preparation and surface measurements. A similar observation was made during the smear measurements. The first circle smear showed about twice the activity of the second, but a third smear 2.5 hours latter showed the surface activity had recovered to its original level. The Lumi-Scint activities for smears 1 and 2 also increased by 10-20% during this time. This is frequently observed for smears, where it may take several hours for all the activity on the paper to leach into the cocktail. A second count at least 10 minutes or so after the first is also a useful check against false readings due to cocktail luminescence.
This same procedure was attempted at 420-D, but the level of activity prevented effective comparisons for all but the electret measurements. Because of the low tritium activity, electret measurements of 24 hours or more were necessary to obtain meaningful readings. Two electret checks, a week apart, were made on a candidate area before any surface preparation. The second reading was about 10% lower than the first. The surface was then prepared by grinding and vacuuming immediately prior to setting out two low-level electrets side-by-side in the work circle (at the grinding location). Two successive 24-hour measurements were made, the latter being about half the first. A third post-grinding measurement was made 5 days later which agreed with the second one. One area in the circle consistently measured about twice the other during all three electret measurements. Two smears of the circle were taken, but the Lumi-Scint was not sensitive enough for these. Instead, readings were only obtainable using a high efficiency laboratory LSCWhat about calibrating the electret?. The fraction of activity on the smears was only about 0.1% of the average from the electrets. This compares to about a 5% fraction for the 234-H study, which is typical of smear/LSC efficiency for tritium contaminated surfaces. Assigning a surface activity for this location, considering the day-to-day variability in the 420-D surface measurements, is covered in the Discussion of Results section.
After the surface measurements were completed, depth profile samples of the underlying bulk were collected. This was accomplished using a hand-held hammer drill with 16-mm and 19-mm diameter bits, in similar fashion to that described in reference [4]. At 234-H, a grout on concrete floor was 10 cm thick and was drilled in 2.5-cm increments. At 420-D, the concrete was 24 cm thick and was drilled in 4-cm increments. Samples were collected by using a small brush to sweep up the drill powder after each increment and immediately transfer the powder to labeled glass vial. Drilling and collecting the powder from each increment took only a minute or two, which results in a negligible loss of tritium from the sample [4]. The drill hole was vacuumed after each increment to remove any remaining powder before the next drill sample was taken.
This method of sample collection has many advantages compared to core drilling including, cost, speed, and simplicity. Samples are easily packaged and transported for analysis, and they can be stored indefinitely with no additional preservation steps. Sample preparation for analysis (see next section) is trivial and results can be available a few days after collection. Since 20 grams of powder or more is collected, and only a few grams are needed for analysis, replicate analyses are easily performed. Drilling is also very clean. Air samples taken while drilling the high-level 234-H samples showed no measurable increase in air activity or contamination to the immediate area around the work circle. Finally, the process causes almost no structural damage and generates little waste at the site or analysis laboratory.
There are a few minor disadvantages. It is difficult to drill depth increments of less than about 2 cm each or to drill deeper than about 60 cm. For characterization work, finer increments or greater depths are unlikely to be necessary. A final drawback is that single location incremental drill samples may not be entirely representative of the bulk, especially for concretes that contain large-sized aggregate. A simple remedy to this, and used in this study, is to drill multiple holes so the analyses from each can be averaged at each penetration depth. Three holes were drilled in each work circle, which seemed to work well with little added time. (If an additional expense of adding calcium to the analysis is acceptable, as will be seen in the results section, a single hole is satisfactory for sampling.)
Sample preparation has been dramatically quickened and simplified in this study compared to the aqueous leach methods described in the literature. Leach methods generally required 1-2 weeks of leaching the samples in water prior to analyzing an aliquot of the leachate by LSC. The method used in this study involves leaching 1 g of drill-powder in 10 mL of 1N HNO3 overnight. Slightly more than 2 ml of the leachate is then distilled, and a 2 ml aliquot of the distillate is analyzed in 18 ml of cocktail for tritium by LSC. The distillation step assures that only tritium is counted even if other radionuclides are present.
To verify the quantitative recovery of tritium by this new method, a comparison was made with a recovery performed using furnace distillation of the drill-powder. This involved heating about 10g of powder in a tube furnace to 550°C for 1-2 hours. Water was swept from the furnace with a flow of dry air into a small condensing flask. The comparison was made for three composite samples collected during the 420-D study. Composites are made up of a portion of all the vacuumed powder collected between profile increments, which should produce a homogeneous sample. Results of the comparison are shown in Table 1.
Sample |
H-3
by Furnace |
H-3
by Leach |
Agreement |
420-D-1 |
5.99 |
6.44 |
7.2 |
420-D-2 |
5.95 |
6.44 |
7.9 |
420-D-3 |
5.81 |
6.26 |
7.5 |
The sample ID numbers ending in 1-3 represent three samples of the combined composite material from all three holes drilled, and the result are in nCi/g of powder analyzed. Several things are apparent from the data. The composite samples are nearly the same and so are apparently representative. Both analysis methods are quite reproducible. And finally, the leach method collects a higher fraction of tritium than the furnace method as used here. This is not surprising in view of Numata's results, which show that temperatures of at least 700°C are necessary to drive off the last traces of water in cement samples (5-10%) [2]. AlsoAlso point out that the remaining fraction is consistent with earlier thermgravimometry., a small loss of water from the condenser in the furnace method is unavoidable, so a low bias for this method is reasonable and expected.
Since water and therefore tritium resides largely in the capillary and gel pores, calcium silicate, and calcium hydroxide phases of hardened cement in concrete; a good correlation of tritium to calcium in samples is anticipated. To test this hypothesis, a calcium analysis was performed on all drill samples. Calcium and silicon content can be useful in distinguishing concretes from grouts, grouts normally having a higher concentration of both elements than concrete. A standard sodium peroxide fusion solubilized the samples, and then the resulting solution was analyzed for Ca and Si by inductively coupled plasma emission spectrometry (ICP-ES). This modern technique is also useful to analyze nearly all other metal species in samples, if these are of interest, and is much simpler and faster than older wet chemical procedures [17].
Results of the tritium, calcium, and silicon analyses for each of the depth profile samples from 420-D are shown in Table 2. The ID number for the samples are given in the first column. The first two fields of the sample ID identify the facility designation, while the last two indicate the sample hole and profile increment number, respectively. The second column lists the approximate depth of the profile increment in centimeters. The third, fourth, and fifth columns list the analyte result in microcuries or as a weight percent of the sample matrix. Column six lists the tritium concentration divided by the calcium content in that profile sample. The last column shows the sums of the of the column-six profile results for each of the three sample holes. Also in their respective columns are the averages and standard deviations of profile results. The averages for the composite samples are shown in the bottom row.
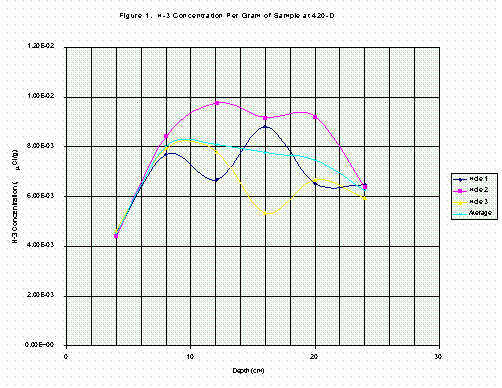
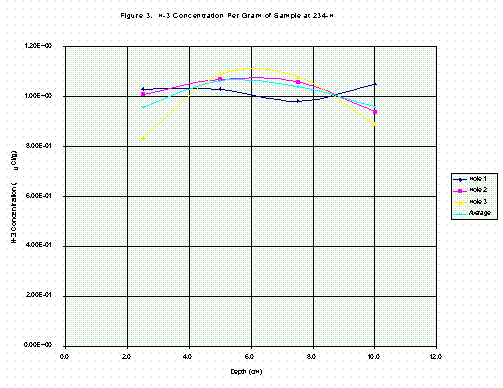
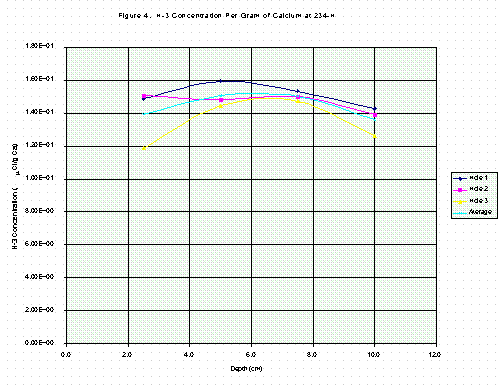
Table 3 contains results for profile samples from 234-H, shown in the same way as Table 2. Comparisons of values in Tables 2 and 3 are both surprising and enlightening. Tritium concentrations in columns three or six, as a function of depth, do not resemble concentration profiles in the literature studies [3, 4, 5, 6]. Instead, the profiles in this study are nearly flat and constant (Figures 1, 2, 3, 4). This is particularly true for the 234-H samples in Table 3 (Figs. 3, 4).
Sample
|
Depth |
H-3 |
Ca |
Si |
H-3/Ca |
Profile
Sum |
420-D-1-1 |
0-4 |
4.55E-03 |
7.31E+00 |
3.00E+01 |
6.22E-02 |
|
420-D-1-2 |
4-8 |
7.69E-03 |
6.87E+00 |
3.23E+01 |
1.12E-01 |
|
420-D-1-3 |
8-12 |
6.68E-03 |
5.58E+00 |
3.23E+01 |
1.20E-01 |
|
420-D-1-4 |
12-16 |
8.80E-03 |
7.33E+00 |
3.20E+01 |
1.20E-01 |
|
420-D-1-5 |
16-20 |
6.52E-03 |
6.16E+00 |
3.12E+01 |
1.06E-01 |
|
420-D-1-6 |
20-24 |
6.47E-03 |
6.15E+00 |
3.15E+01 |
1.05E-01 |
6.25E-01 |
420-D-2-1 |
0-4 |
4.41E-03 |
7.86E+00 |
3.00E+01 |
5.61E-02 |
|
420-D-2-2 |
4-8 |
8.42E-03 |
6.08E+00 |
2.98E+01 |
1.38E-01 |
|
420-D-2-3 |
8-12 |
9.75E-03 |
6.25E+00 |
3.09E+01 |
1.56E-01 |
|
420-D-2-4 |
12-16 |
9.17E-03 |
6.06E+00 |
3.10E+01 |
1.51E-01 |
|
420-D-2-5 |
16-20 |
9.21E-03 |
6.08E+00 |
3.13E+01 |
1.51E-01 |
|
420-D-2-6 |
20-24 |
6.37E-03 |
5.06E+00 |
3.09E+01 |
1.26E-01 |
7.79E-01 |
420-D-3-1 |
0-4 |
4.63E-03 |
6.85E+00 |
3.06E+01 |
6.76E-02 |
|
420-D-3-2 |
4-8 |
7.93E-03 |
6.59E+00 |
3.09E+01 |
1.20E-01 |
|
420-D-3-3 |
8-12 |
7.79E-03 |
7.29E+00 |
3.01E+01 |
1.07E-01 |
|
420-D-3-4 |
12-16 |
5.32E-03 |
4.99E+00 |
3.00E+01 |
1.07E-01 |
|
420-D-3-5 |
16-20 |
6.67E-03 |
6.78E+00 |
2.87E+01 |
9.84E-02 |
|
420-D-3-6 |
20-24 |
5.92E-03 |
7.26E+00 |
2.92E+01 |
8.15E-02 |
5.81E-01 |
Average |
7.02E-03 |
6.48E+00 |
3.07E+01 |
1.10E-01 |
6.62E-01 | |
Std.Dev. (%) |
2.39E+01 |
1.23E+01 |
3.27E+00 |
2.68E+01 |
1.57E+01 | |
420-D |
Composite |
6.38E-03 |
6.23E+00 |
3.05E+01 |
1.02E-01 |
The top, 0-4 cm, tritium profile samples for all three holes at 420-D are each about 40-50% lower than the otherwise flat average. This is likely caused by weathering effects of an outdoor environment on the near surface material. The frequent wetting and drying from rain water is expected to dilute and exchange with any tritiated water in the concrete from earlier exposures to moderator storage tank leaks. This effect would be most pronounced at or near the top surface, as the data show. In addition, surface cracking from weathering could allow additional surface area for water to move between the environment and the concrete.
The calcium values for the 420-D concrete show a standard deviation in the average of almost 12% while that for the 234-H grout is only 5%. The lack of large aggregate, which is usually calcium-free, in the grout implies the material is more homogeneous, with a nearly constant calcium content compared to concrete. The average calcium and silicon values in Tables 2 and 3 appears to be slightly high compared to estimated typical concrete and grout mixes used at SRS (Table 4). However, the analysis values in Table 2 and 3 are based on dry weight, while Table 4 values are calculated assuming no water loss.
Agreement between the composite samples and the group profile averages for calcium and silicon are representative of the precision in these analyses. This is not the case for tritium because of its volatility. In vacuuming the powder debris, the material is bathed in a strong air stream as both powder and air are pulled into the vacuum cleaner's collection bag. The tritium loss was relatively small for the 420-D samples which were collected in a time span of less than an hour, but was much larger for 234-H where a delay in completing sampling left the composite sample powder in the vacuum cleaner exposed to ambient air overnight.
Sample
|
Depth |
H-3 |
Ca |
Si |
H-3/Ca |
Profile
Sum |
234-H-1-1 |
0-2.5 |
1.03E+00 |
6.93E+00 |
3.40E+01 |
1.49E+01 |
|
234-H-1-2 |
2.5-5.0 |
1.03E+00 |
6.46E+00 |
3.58E+01 |
1.59E+01 |
|
234-H-1-3 |
5.0-7.5 |
9.82E-01 |
6.41E+00 |
3.60E+01 |
1.53E+01 |
|
234-H-1-4 |
7.5-10.0 |
1.05E+00 |
7.36E+00 |
3.40E+01 |
1.43E+01 |
6.04E+01 |
234-H-2-1 |
0-2.5 |
1.01E+00 |
6.71E+00 |
3.30E+01 |
1.51E+01 |
|
234-H-2-2 |
2.5-5.0 |
1.07E+00 |
7.23E+00 |
3.36E+01 |
1.48E+01 |
|
234-H-2-3 |
5.0-7.5 |
1.06E+00 |
7.07E+00 |
3.44E+01 |
1.50E+01 |
|
234-H-2-4 |
7.5-10.0 |
9.39E-01 |
6.75E+00 |
3.49E+01 |
1.39E+01 |
5.88E+01 |
234-H-3-1 |
0-2.5 |
8.34E-01 |
7.02E+00 |
3.41E+01 |
1.19E+01 |
|
234-H-3-2 |
2.5-5.0 |
1.09E+00 |
7.55E+00 |
3.33E+01 |
1.44E+01 |
|
234-H-3-3 |
5.0-7.5 |
1.08E+00 |
7.33E+00 |
3.16E+01 |
1.47E+01 |
|
234-H-3-4 |
7.5-10.0 |
8.90E-01 |
7.04E+00 |
3.35E+01 |
1.26E+01 |
5.37E+01 |
Average |
1.01E+00 |
6.99E+00 |
3.40E+01 |
1.44E+01 |
5.76E+01 | |
Std.Dev. (%) |
7.98E+00 |
5.09E+00 |
3.53E+00 |
7.89E+00 |
6.06E+00 | |
234-H |
Composite |
4.86E-01 |
6.91E+00 |
3.21E+01 |
7.03E+00 |
Material |
Water/Cement |
Sand/Cement |
Stone/Cement |
Calcium |
Silicon |
Concrete |
0.55 |
3.0 |
5.0 |
5.0 |
31.4 |
Grout |
1.0 |
5.0 |
0 |
6.4 |
34.7 |
The significance of the sixth column in Tables 2 and 3 is illustrated by comparing Fig. 1 with Fig. 3 and Fig. 2 with Fig. 4. The depth profiles for tritium per gram of sample are plotted and show considerable fluctuation in Fig. 1 for 420-D but somewhat less in Fig. 3 for 234-H. In Figs. 2 and 4, the tritium per gram of calcium is plotted and is seen to be less variable, especially in Fig. 2. By expressing the tritium concentration relative to the amount of calcium in the sample, the effects of drilling through stone aggregate, which contains no tritium and low calcium, are minimized. The difference in variability is much less pronounced for the grout material sampled at 234-H as shown in Fig. 3 and 4, since grout contains only sand and cement as solids and is much more homogeneous than concrete. The connecting lines in Figs. 1-4 are for clarity only.
Fig. 2 also shows that the tritium concentrations for the Hole 2 profiles are quite a bit higher than for those of Hole 1 or Hole 3. This is consistent with the factor of two difference observed in the electret measurements, as Hole 2 was located nearer the high reading electret. Such local concentration differences probably are not unusual because of the uneven nature of liquid spills.
Comparing Figs. 2 and 4 shows that both areas have similar profile distribution shapes: i.e. nearly homogenous concentration in the bulk of the material except for the somewhat lower 0-4 cm increments at 420-D noted earlier. This similarity This is very important!!!! And I think you're absolutely correct!strongly indicates that at both locations the contamination mechanism was the same. Because the surface at 420-D was certainly exposed to liquid DTO, 234-H must also have been exposed to liquid. The source of liquid contamination in 234-H, which processes elemental tritium was, at first, unclear. But it was later learned that highly contaminated fluids (pump oil, etc.) were stored in the process cabinet near the study location, and that spills occasionally happened. This fluid was able to leak out of the cabinet and onto the adjacent floor. Cleanup involved mopping which likely served to further spread contamination about the floor. Tritium concentrations for Hole 3, by comparison of the profile-sum data of Table 3, may be somewhat low with respect to the other two. This also suggests a liquid mode of contamination as opposed to one resulting from gaseous elemental tritium releases into a room, where facility ventilation would rapidly mix the contaminant to a fairly uniform concentration over small areas of exposed floor.
The above results clearly support the expectation of tritium behavior in hydrated cement paste and concrete as outlined earlier. At the 420-D area, the concrete surface was exposed to decades of sporadic accidents of wetting by liquid DTO with tritium concentrations as high as 5-10 mCi/mL. The surface was exposed to rain water in between contamination incidents, but there likely was no hydraulic flow to flush the tritiated water out of the capillaries-added water to the capillaries would only dilute any free tritiated water remaining. The net result of these events is that tritiated water became integrated into the concrete with time, including possible further cement hydration. Diffusion and exchange of the tritium contaminant with other hydrogen species throughout the concrete has led to a near uniform distribution, except at the top surface which continues to be perturbed and depleted more quickly from weather exposure. This happens frequently enough in the moist climate at SR that diffusion and exchange of tritium from the lower depths probably cannot maintain an equilibrium concentration with the near surface region.
The scenario of events at the 234-H location is slightly different. From discussions with personnel familiar with past operating conditions, it is apparent that exposure to liquids containing 10-50 times more tritium were possible. Although the frequency and identity of the fluid contamination is unknown, the net result was much the same as at 420-D. Once the relatively dry grout in this climate-controlled indoor facility was exposed to a surface spill of liquid, surface capillaries would have filled rapidly. This would be followed by tritium exchange with water species in the grout, and a much slower diffusion into the bulk, leading eventually to a nearly uniform distribution. The only two modes of tritium release out of the bulk would be by fast evaporation of capillary water and the much slower diffusion from the solid. In an indoor and environmentally controlled room such as this, perturbations to a slow, steady diffusion are unlikely. And the nearly flat tritium concentration profile observed is consistent with this.
Figure 5 is a comparison of the bulk tritium levels obtained from the two liquid-contaminated study areas in this report, with what is typically found in facilities contaminated by airborne exposures (estimated from data in reference [2]). The tritium concentrations in the contamination media are rough estimates based on process knowledge and operations in the respective facilities. In all three cases, the depth profiles were obtained by hammer drilling, with the drill powder leached and analyzed for tritium. The values at the surface are extrapolations from the first profile sample in each case. The contrast in profile shapes for the upper two curves and the lower one is very apparent. The liquid contamination mode, clearly, produces a relatively flat profile; and one that can hold very high levels of tritium when compared to that produced by the airborne mode. The difference in tritium concentration and density between air and liquids determines and dominates the profile.
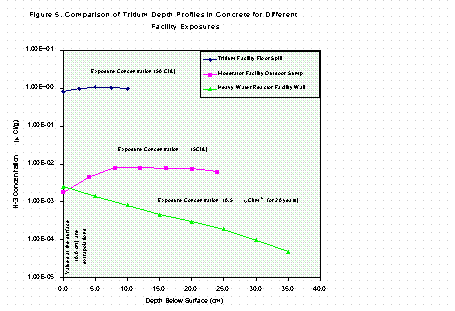
It is reasonable to ask whether the measured high tritium concentrations arising from a liquid spill are credible. From evaporable water depth profiles in reference [11], a rough estimate of the amount of water that might quickly be sorbed into the upper surface capillaries is about 0.05 mL/cm2 under ambient air conditions. The tritium level in the moderator water responsible for the contaminated sump at the 420-D facility was close to 5 mCi/mL. So, up to 250 µCi of tritium per square centimeter of surface area might have been available to permeate into the underlying bulk. This amount distributed uniformly through the 24-cm slab thickness would give a concrete concentration of about 5 µCi/g. From Table 2, the average value measured was only 7 nCi/g, or nearly a factor of a thousand times less. The question instead should be, "Why is the concentration so low?"
As mentioned earlier, the only modes of tritium release from the bulk material is from capillary evaporation or diffusion. Because diffusion is a slow continual process, fast evaporative loss is probably mostly responsible. Immediately after a spill the surface will again begin to dry. In a matter of a few days most of the water sorbed into the surface capillaries will again dry to the air, or perhaps be diluted by rain. In this short time, diffusion and exchange mechanisms will not have progressed very far. The 234-H tritium facility has an average measured bulk value (see Table 3) of 1 µCi/g. By analogous calculation, this is lower than the maximum calculated value by a factor of one hundred. That this difference is less than at 420-D may be from the absence of rain and repeated wetting and drying. However, here the tritium concentration of the exposing liquid is much less certain, so no firm conclusion can be reached.
A further difference between airborne and liquid exposures of concretes and cements is in the speed at which the tritium permeates the bulk. Comparing the 420-D concrete sump to the heavy water reactor wall, the difference in the tritium concentrations in the contaminating media is ten orders of magnitude. Although the former case is one of liquid diffusion and the latter is one of gaseous diffusion, at most the offset in their respective diffusion coefficients is perhaps five orders of magnitude, (see discussion in following section). Other things being equal, initial progression of the diffusion front could be 100,000 times faster for the liquid exposure compared to the gaseous one. This would be reduced by perhaps a factor of 1000 quickly by evaporative lose, but the liquid front can still be expected to move very fast compared to the gas.
It is also apparent from the extrapolated surface activities, that surface measurements alone are not sufficient to predict the profile shape.
One of the primary objectives of this study was to determine whether measurements of tritium contamination on the surface of concrete/hydrated cement can be related to that in the bulk. Various studies by Numata and others have shown that the uptake of HT and gaseous HTO species in cement and concrete can be described by Fick's laws of diffusion. Here the instantaneous flow per unit area of the tritium species either into or out of the bulk is equal to a suitable diffusion coefficient times a concentration gradient. There are no instruments capable of directly measuring such a flow. Testing instead has been based on mass transfer studies where the fraction of tritium released from the bulk can be related to time from solutions of appropriate differential equations and experimental boundary conditions. One approximation commonly used is a semi-infinite plane source diffusion model [18, 19]. With it Masuzuru [7] measured effective diffusivities of 3.6 ± 3.3 x 10-9 cm2/s using deionized water to leach tritium from a variety of hardened cement pastes. These may be contrasted with typical values of 10-4-10-5 cm2/s for the self diffusion of tritiated water, and 100-10-2 cm2/s for the gaseous self diffusion of water vapor. Thus the effective diffusion of tritiated water within hardened cement pastes is an extremely slow process in comparison.
None of the various literature models discuss the problem of physically measuring the amount of tritium activity on a cement surface and correlating it to that in the underlying bulk. To do so, the specifics of the actual surface activity measurement must be considered. As was already discussed, the traditional approach of smears combined with LSC is very sensitive, however also quite variable and subject to many uncontrolled environmental and operator factors. Comparing smear values to those from the SAM and from electrets suggest that about 5% of the surface activity was picked up by a smear at 234-H but only about 0.1% at 420-D, a factor of 50 difference. The problem is that a smear can only pick up transferable moisture or contaminated particles on surfaces.
Both the SAM and electret work on the ionization chamber principle of measuring the radiation produced by contamination on or very near the surface [20]. They differ only in the way they measure the ion current and in their relative sensitivity to tritium. Although the tritium betas are very low in energy, they still travel a finite distance producing ionization along their path. In air that path is at most about 0.5 cm [20]; in water it is about 6x10-4 cm [21]. A fundamental question related to this work is: what is the tritium beta range in cement?
At first thought, the tritium beta range in hydrated cement, having density ~2 g/cm3, would seem to have to be less than that in water. However, it is the areal density not the bulk density which is of concern at the surface. From references [12] and [13], hardened cement has a specific surface area of about 2x106 cm2/g for an implied areal density of about 5x10-7 g/cm2. This very small areal density means that tritium betas will lose little energy as they leave the concrete/cement surface and might have a range somewhere between the bounding extremes of air and water. From the measurements taken at the two study locations, the high concentrations for 234-H, which could be measured independently with the SAM and electrets, are likely to be the most reliable. From Table 3, the average bulk activity was analyzed as about 1 µCi/g, and the measured surface activity was about 14 nCi/cm2. The correlation factor between the two is simply the surface activity divided by the bulk activity (14 nCi/cm2/1000 nCi/g) or 1.4x10-2 g/cm2. Dividing this value by the approximate bulk density of the material gives the beta range as about 7x10-3 cm, which is an order of magnitude greater than that in water. This is equivalent to every square centimeter of surface area appearing as a thin disk having a volume of 7x10-3 cm3.
The cement material at 234-H is mostly a grout. Because concrete is typically about 15-20% more dense than concrete, a correlation factor for 420-D might be estimated as about 1.2x10-2 g/cm2. Using this value, Table 5 shows the comparison results for both study areas. The agreement between the estimated and measured surface activities is surprisingly good.
Study |
Bulk
Mass |
Correlation |
Surface
Activity |
Surface Activity |
420-D |
7.0 E-03 |
1.2E-02 |
0.8E-01 |
0.2 - 4.1 E-01 |
234-H |
1.0 E+00 |
1.4E-02 |
1.4E+01 |
1.4 E+01 |
This is certainly not an extensive comparison, particularly in view of the 20-fold range for the 420-D measured surface activity. The outdoor location and rain or other weather effects are the likely causes of this variation in measured surface activity. For all the 420-D surface measurements, as chance would have it, rain had occurred just prior to or during the time the electrets were deployed. Only some months later was the effect of rain evident on electrets measurements. Another electret measurement at this location was made on 9/15-9/17/98, two days during an extended period of hot weather and no rain. The surface activity was measured again as 0.02-0.06 nCi/cm2. The electret was placed about 10 cm from the previous work circle which had been grouted-over following the drilling work in May. Because of this difference in placement, the value may not exactly represent that obtained in the earlier work, but it certainly tends to support the lower range shown in Table 5. A third electret measurement was made from 1/12-1/18/99. Two electrets were used: one on a plastic plate to block tritium surface activity and the other directly on the surface. Both were placed side by side in the old grouted work circle with a cover over the top to protect the electrets from rain. A heavy rain happened to occur on 1/17-1/18/99. Although this did not wet the electrets themselves, the surface underneath both was thoroughly wetted.
The implied surface activities from the discharge rates of the electrets were 9x10-3 nCi/cm2 for the electret over the plastic plate and 0.35 nCi/cm2 for the one over the grouted concrete surface, which is nearly the same as the original activity measured nine months earlier. But the surface had been grouted over, and no processing or leaks occurred in the intervening months. So, it is unlikely that this new result is entirely due to tritium. It is more likely that the high value is the result of radon in the rain water which wetted the grouted surface on the last day of the measurement. The washout of atmospheric radon in rain is a very noticeable background-increasing effect in many kinds of radiation counting work. For instance, outdoor conveyor-belt counting systems often have to suspend operation due to background increases even in light rains. Once all surface moisture evaporates, backgrounds decrease and operation can resume. In the case of a cement surface, with its ability to imbibe water, the elevated background may persist for several days after a rain as some of the activity would persist from 3.8-day Rn-222 progeny produced in the natural U-238 decay chain. The factor of 40 lower background measured by the electret on the plastic plate is likely representative of the gamma-ray or non-surface radiation background effects.
It is interesting to note that the results from the smears taken at the 420-D site may actually be close to a true indication of the tritium surface activity after all. As mentioned above, smears counted on a very sensitive laboratory LSC showed only about 0.1% of the activity indicated by the electret measurements at that time. No activity other than tritium was present, but this was many days after the smears were made. Any radon activity on the smears would have decayed away in that time. Using a collection efficiency of 5%, roughly typical of smears for tritium, the actual surface activity is computed as 0.01 nCi/cm2, again close to the lower range in Table 5. In view of these various effects on the measured range, the average value of about 0.2 nCi/cm2 is probably a reasonable estimate of the actual tritium surface activity at the 420-D location.
The average correlation factor from Table 5 is 1.3x10-2 g/cm2. Dividing the measured surface activity of a cement surface by this value will give the approximate bulk concentration of the immediate underlying material. This is the number that is of interest in D&D work because it determines how to the material must be disposed of and at what cost. This can only be done where the tritium activity profile is reasonably constant. Unfortunately, surface activity alone cannot distinguish between a diffusion profile resulting from gaseous exposure of the cement, and the nearly constant profile produced from liquid exposures. The exposure mode is obvious for water filled structures, such as reactor fuel storage basins or process equipment sumps. But even facilities which process or handle only elemental tritium may have localized areas where liquid exposure has occurred. Such areas may hold much more tritium than those subjected to gaseous exposures.
Still, surface measurements for cementitious materials suffering gaseous exposures can be used to give reasonably good estimates of the bulk tritium burden. Many studies have shown that the diffusion distribution of tritium in concrete can be fit with theory-based models to good accuracy [4, 6, 22]. Results depend, as one might expect, on assumed diffusion coefficients and porosity values, but these usually can be estimated to within a factor or two or three. With further estimates, as to the duration of exposure and concentration gradient, a reasonable depth profile can be calculated.
Sampling the cement mass by hammer-drilling is so quick, easy, and structurally harmless, that it should always be done in D&D activities, at least on a limited basis, to verify the suspected distribution. If the exposure was diffusion controlled, such a profile will likely be needed to determine if and how much of the top surface should be removed during D&D. Surface activity surveys are useful in locating tritium contamination regardless of the exposure mode, but only when the depth profile is known can plans for cleanup or disposal be realistically made. For either exposure mode, the maximum levels of tritium contamination will be found near the topmost layer of the cement surface exposed, although heavy or extended weathering and diffusion will slightly reduce the surface amount in the absence of new exposure.
Surface tritium and bulk tritium activity were measured using several methods at two locations at the Savannah River Site. These data, along with information in the literature, resulted in the following conclusions.
Two new methods of in-situ measuring of tritium surface activity were successfully tested: the Surface Area Monitor and the electret. These should enormously simplify the characterization of tritium in concrete/cement materials especially for large basin structures or facilities known or suspected to have large tritium inventories. In addition, a quick and easy method of depth-profile sampling of cement and concrete using hammer drilling was successfully demonstrated. A new nitric acid leaching method, which allows tritium measurement in the presence of other activities, was also developed and demonstrated.
Depending on whether the exposure occurred via a liquid or a gaseous mode, the variation of bulk activity with depth will likely differ. From numerous examples in the literature, gaseous exposure to either the elemental or oxide forms of tritium invariably leads to a diffusion profile where most of the activity is at the exposed surface and decreases rapidly with depth into the bulk. This study revealed a previously unreported tritium distribution occurs for liquid exposure modes. Here the profile is largely constant except for some lowering at the top surface, and perhaps to a lesser extent at other boundary surfaces, from diffusion and atmospheric weathering.
Surface and bulk tritium contamination in concrete/cements were observed to be directly proportional to one another, if the tritium contamination resulted from exposure to contaminated liquid, and if the surface activity is measured from the resulting ions created in air above the surface. An intuitive model based on the effective surface area of concrete was created that explains the surface to bulk relation.
Finally, the results this study, as well as those involving gaseous exposure, can be understood in terms of the physical and chemical nature of concrete. This is important and informative in understanding how and where water is distributed in hardened cements and what occurs if the concrete is wetted again. It also forms the basis for a physical model of what a cement surface measurement sees and how this can be related to tritium in the bulk.