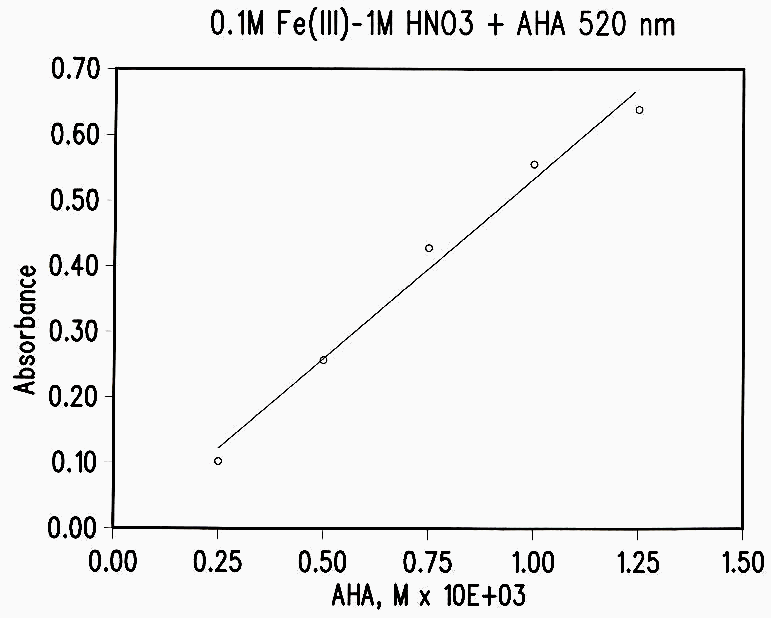
Figure 1 Standard Curve
WSRC-TR-2002-00283
Radiation Chemistry of Acetohydroxamic
Acid in the UREX Process
D. G. Karraker
Westinghouse Savannah River Company
Aiken, South Carolina 29808
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
This report has been reproduced directly from the best available copy.
Available for sale to the public, in paper, from: U.S. Department of Commerce, National Technical Information Service, 5285 Port Royal Road, Springfield, VA 22161, phone: (800) 553-6847, fax: (703) 605-6900, email: orders@ntis.fedworld.gov online ordering: http://www.ntis.gov/help/index.asp
Available electronically at http://www.osti.gov/bridge
Available for a processing fee to U.S. Department of Energy and its contractors, in paper, from: U.S. Department of Energy, Office of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831-0062, phone: (865 ) 576-8401, fax: (865) 576-5728, email: reports@adonis.osti.gov
Introduction
The UREX process is being developed to process irradiated power reactor elements by dissolution in nitric acid and solvent extraction by a variation of the PUREX process.1 Rather than recovering both U and Pu, as in Purex, only U will be recovered by solvent extraction, hence the name "UREX." A complexing agent, acetohydroxamic acid (AHA), will be added to the scrub stream to prevent the extraction of Pu(IV) and Np(VI). AHA (CH3C=ONHOH) is decomposed to gaseous products in waste evaporation, so no solid waste is generated by its addition.
AHA is hydrolyzed in acid solution to acetic acid and hydroxylamine at a rate dependent on the acid concentration.2-4 The fuel to be processed is ca 40 years cooled, 30,000-50,000 MWD/MT material; although only a few fission products remain, the Pu isotopes and 241Am generate a radiation field estimated to be 2.6E+02R during processing. (see Appendix for calculation.) This study was conducted to determine the effect of this level of radiation on the stability of AHA during processing.
Summary
The irradiation of AHA by 60Co gammas to doses up to 1.1E+06 R/hr found that radiation decomposed a minor fraction of AHA compared to the loss by hydrolysis. The radiolysis had a weak dependence on acid concentration and a weak dependence on total dose; a factor of 5 increase in dose decreased the AHA concentration by less than a factor of two. It is proposed that this effect is due to scavenging of radiation-produced species by the hydrolysis products of AHA, acetic acid and hydroxylamine. Radiation will not be a significant factor for AHA use in the UREX process.
Experimental Section
Reagents
AHA was purchased from Aldrich Chemical Co. as 98% pure and was used without any further purification. Nitric acid, acetic acid, and ferric nitrate were C.P. grade. Hydroxylammonium nitrate (HAN) was obtained from the PUREX plant at the Savannah River Site. Distilled water was used without purification.
Irradiations
Solutions of AHA, HNO3, NaNO3, etc. were irradiated in a 60Co source at dose rates between 9.2 and 8.86 E+05 Rad/hr. The solutions were contained in glass vials that had been heated to 500°C for 2 hours to destroy any organic material present. No further precautions were taken to avoid trace material that might cause unusual effects. Control samples were retained to allow comparison of the radiation effects, as distinct from the loss of AHA from hydrolysis.
Analyses
AHA was analyzed spectrophotometrically by the absorption of the Fe(III)-AHA complex at 505-515 nm. The general procedure was to add a 25-100 µL aliquot of an AHA solution to 10 mL of 1M HNO3-0.1M Fe(NO3)3 and determine the absorbance immediately with an H-P diode array spectrophotometer. The standard curve is shown in Figure 1. It is essential that the determination be made within a few minutes, since the Fe(III)-AHA complex begins to hydrolyze after 5 to 10 minutes.
AHA hydrolyzes in acid solutions at a rate depending on the acidity, as
CH3C=ONHOH + H+ + H2O -à CH3COOH + NH3OH+
(see Figure 2). Unirradiated control samples were analyzed to determine the effect of radiation as distinct from the loss of AHA by hydrolysis.
A few irradiated samples were totally hydrolyzed by adjusting to 2M HNO3 and analyzed by ion chromatography (IC) and gas chromatography-mass spectroscopy (gc-ms) after a delay of a few days to allow complete hydrolysis.
Results
The results of a typical irradiation are shown in the following table.
Table 1 Experimental Results, Dose =7.59E+05 R
|
Solution * |
Absorbance of Fe(III)-AHA |
||||||
|
Control |
Irradiated |
Difference |
|||||
|
0.44M H+ |
0.407 |
0.315 |
0.092 |
||||
|
0.44M H+-0.6M NaNO3 |
0.407 |
0.312 |
0.091 |
||||
|
0.86M H+ |
0.292 |
0.162 |
0.130 |
||||
|
1.08M H+ |
0.201 |
0.093 |
0.108 |
||||
*All samples 0.1M AHA. At make-up, the absorbance of the Fe(III)-AHA complex is about 0.6
The destruction of AHA by radiation is smaller than the loss by hydrolysis during the time between make-up, irradiation and analysis. The loss of AHA by radiation shows a weak dependence on the acid concentration (see Figure 3) but no effect for nitrate concentration.
The effect of dose was also small. The increase in AHA loss as the dose was increased was much less than the expected linear dose dependence (see Figure 4 and Table 2).
Table 2 Effect of Radiation on 0.1M AHA Solutions
|
Absorbance of Fe(III)-AHA Complex* |
||||||||||||||
|
Solution |
Control |
Radiation Dose, R |
||||||||||||
|
1.91E+05 |
3.81E+05 |
7.59E+05 |
1.14E+06 |
|||||||||||
|
0.2M H+ |
0.413 |
0.312 |
0.324 |
|||||||||||
|
0.407 |
0.315 |
0.305 |
||||||||||||
|
0.463 |
0.382 |
0.358 |
||||||||||||
|
0.4M H+ |
0.276 |
0.185 |
0.174 |
|||||||||||
|
0.292 |
0.162 |
0.135 |
||||||||||||
|
0.5M H+ |
0.246 |
0.144 |
0.133 |
|||||||||||
|
0.201 |
0.0093 |
0.080 |
||||||||||||
*1/400 dilution into 0.1M Fe(III)-1M HNO3
The IC analysis of hydrolyzed, irradiated solutions found only acetic acid; gc-ms found peaks at mass 46 and 62 in 0.12 mg/L concentration. The peak at 46 corresponds to formic acid (HCOOH) and the peak at 62 corresponds to a trace of nitrate ion.
Discussion
AHA has sufficient stability toward radiation for its use as a reductant/complexant in the UREX process. There are several possible causes for its deviation from a linear dose dependence. The leading possibility is the scavenging of the primary radiolysis products (OH, H, e-, etc.) by the acetic acid and HAN produced by hydrolysis. As more AHA hydrolyzes, the concentration of acetic acid and HAN increases, and the scavenging becomes more effective. This explanation was adopted more or less because other credible explanations could be disproved experimentally, and this one could not.
Another possibility is the radiolysis of AHA to formohydroxamic acid (HC=ONHOH, FHA). FHA complexes Fe(III) in the same manner as HAN and would be indistinguishable in the analysis. However, only a trace of formic acid was found by gc-ms. The radiolysis of AHA to FHA does not take place to the extent necessary to explain the dose dependence.
The last possibility considered was a radiation-induced back-reaction between acetic acid and HAN to produce AHA. Irradiations of HAN, acetic acid and HNO3 solutions did not produce any detectable AHA.
References
Figure 1 Standard Curve
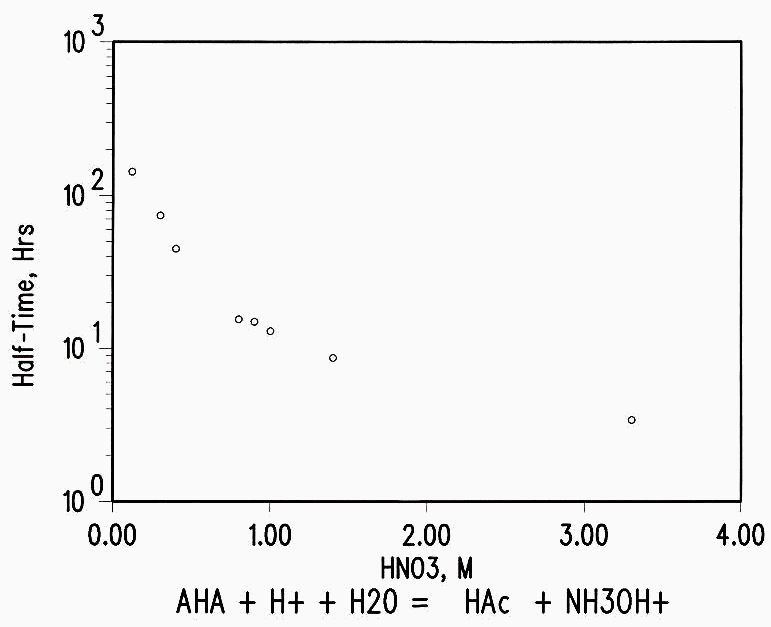
Figure 2 AHA Hydrolysis Rate
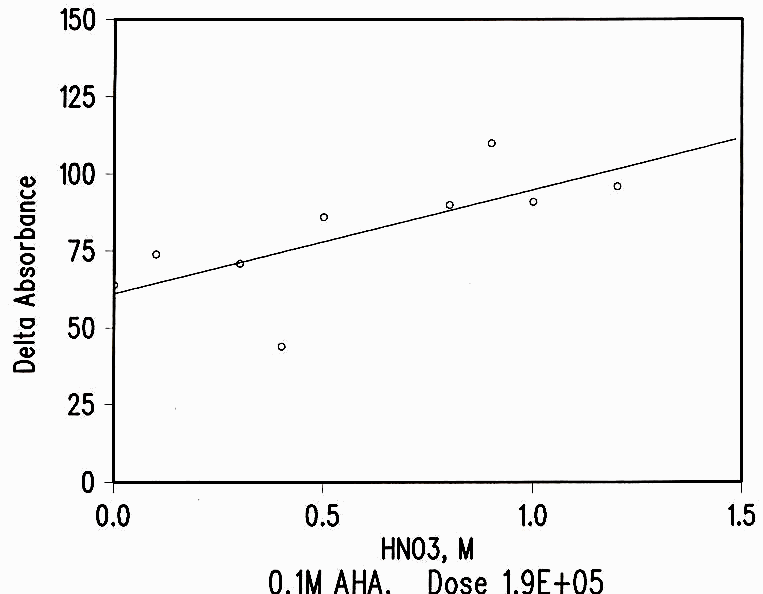
Figure 3 Acid Dependence of Radiation Damage
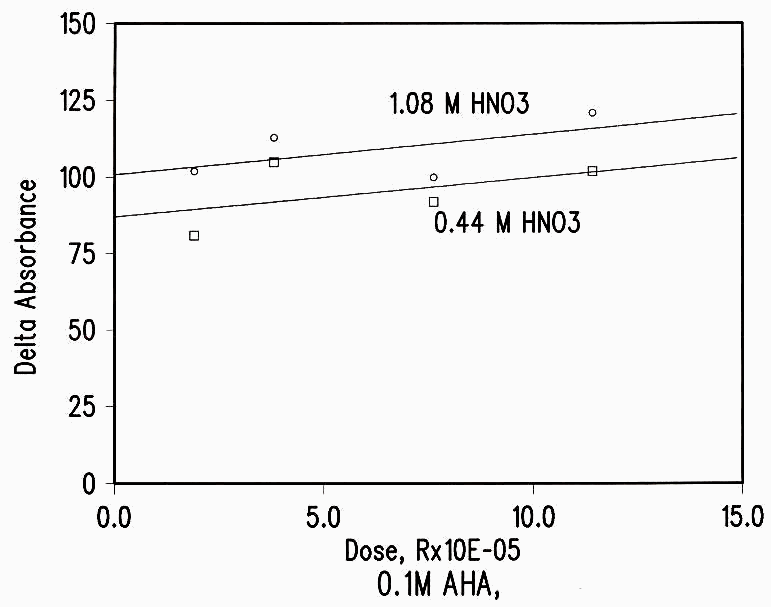
Figure 4 Dose Dependence of Radiation Damage
Appendix Calculation of Radiation Dose
The calculation assumes the following isotopic distribution1 for power reactor fuel irradiated to 30,000 MWD/MT.
|
Isotope |
% At Discharge |
% After 40 yr Decay |
mCi/g x 1.6 g/L |
|
|
238 |
1.3 |
1.1 |
319 |
|
|
239 |
61.3 |
72.6 |
73 |
|
|
240 |
13.1 |
15.5 |
57 |
|
|
241 |
17.7 |
3.0 |
5220 (beta) |
|
|
242 |
6.6 |
7.8 |
0.5 |
|
|
241 Am |
0.15g/gPu |
847 |
||
The isotopic composition was converted to mCi/g, multiplied by 1.6 g/L (1AF flow 100 @ 2.7g/L, diluted by 1AS flow 60) and the radiation dose2 calculated for each isotope from
Dose(R/sec)=0.5927 x C x E
where C is mCi/g and E is Mev/disintegration, taken to be 5.4 Mev for alpha emitters and 0.07Mev for 241Pu. The density of the combined 1AS and 1AF streams was assumed to be 1 g/mL. For the alpha emitters, the sum of the mCi/g values is 1.3 mCi/g and the calculated dose is 4.2 R/sec; for the 5220 mCi/g beta emitter, the calculated dose is 0.2 R/sec. The total dose of 4.4 R/sec exposed in the 10 extraction stages, at 6 sec per stage is 2.6E+02 R.
The calculation does not include Cm isotopes, 243Am and fission products. The calculation has considerable uncertainty because the 238Pu content is not considered reliable. Since it is a major contributor to the dose, its uncertainty is a major source of error.