WSRC-MS-2001-00007
Cerium as a Surrogate in the Plutonium Immobilized Form
J. C. Marra, A. D. Cozzi, R. A. Pierce, J. M. Pareizs,
A. R. Jurgensen, and D. M. Missimer
Westinghouse Savannah River Company
Aiken, SC 29808
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
This report has been reproduced directly from the best available copy.
Available for sale to the public, in paper, from: U.S. Department of Commerce, National Technical Information Service, 5285 Port Royal Road, Springfield, VA 22161, phone: (800) 553-6847, fax: (703) 605-6900, email: orders@ntis.fedworld.gov online ordering: http://www.ntis.gov/support/ordering.htm
Available electronically at http://www.osti.gov/bridge/
Available for a processing fee to U.S. Department of Energy and its contractors, in paper, from: U.S. Department of Energy, Office of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831-0062, phone: (865 ) 576-8401, fax: (865) 576-5728, email: reports@adonis.osti.gov
Abstract
The Department of Energy (DOE) plans to immobilize a portion of the excess weapons useable plutonium in a ceramic form for final geologic disposal. The proposed immobilization form is a titanate based ceramic consisting primarily of a pyrochlore phase with lesser amounts of brannerite, rutile, zirconolite, vitreous phases and/or other minor phases depending on the impurities present in the feed. The ceramic formulation is cold-pressed and then densified via a reactive sintering process. Cerium has been used as a surrogate for plutonium to facilitate formulation development and process testing. The use of cerium vs. plutonium results in differences in behavior during sintering of the ceramic form. The phase development progression and final phase assemblage is different when cerium is substituted for the actinides in the form. However, the physical behavior of cerium oxide powder and the formation of a pyrochlore-rich ceramic of similar density to the actinide-bearing material make cerium an adequate surrogate for formulation and process development studies.
Introduction
The U.S. Department of Energy (DOE) has determined that at least a portion of the excess weapons-useable material will be immobilized in a titanate-based ceramic for final disposal in a geologic repository [1]. The technology to be employed will involve immobilizing the Pu in ceramic "pucks", placing the pucks in cans and then encasing these cans in radioactive high-level waste glass (i.e. can-in-canister) [2]. The can-in-canister configuration, including the high radiation field afforded by the waste glass, will provide the necessary proliferation resistance.
The proposed immobilization formulation contains chemical precursors to produce a durable ceramic form as well as sufficient neutron absorbers to preclude criticality. The baseline formulation shown in Table I was designed to produce a ceramic consisting primarily of a highly substituted pyrochlore with minor amounts of brannerite and hafnia-substituted rutile. Many of the prospective feed streams contain large amounts of uranium in addition to plutonium. The pyrochlore phase was shown to readily accommodate large quantities of actinides [3]. In addition, given that pyrochlore has a cubic structure, it is anticipated that the isotropic nature of the mineral phase will minimize radiation damage [3]. When impurities are present in the Pu feed-streams other minor phases such as zirconolite, perovskite, glassy phases, and/or other minor phases may be present in the ceramic form. The composition was designed such that all of the phases that contain actinides also have significant amounts of neutron absorbers (with the exception of unreacted actinide oxide phases - present at less than 1 vol %). Chemical formulas (showing the atomic substitutions) for the various mineral phases observed in the ceramic are shown in Table II.

Concurrently with formulation development efforts, a ceramic fabrication process is being developed based on cold pressing and sintering. In summary, the current baseline process consists of:
Nominal sintering conditions have been established for the ceramic form consisting of heating at 3°C/min to 300°C, 2 hour hold at 300°C for binder burnout, 5°C/min to 1350°C, hold at 1350°C for 4 hours, and cooling at 5°C/min to room temperature [3]. The objective of the sintering process is to produce a monolithic product with a phase assemblage that will ensure product durability. Unlike solid state or single phase sintering processes, the sintering of the Pu immobilized ceramic form is "reactive" in nature, where the oxide precursors react to form highly substituted mineral phases. Cerium has been used as a surrogate for plutonium to facilitate formulation development and process testing. The use of cerium vs. plutonium results in differences in behavior during sintering of the ceramic form. The objectives of this work are to quantify these behavioral differences to determine the adequacy of cerium as a surrogate for the actinides in the ceramic form.
Experimental
Precursor Batch and Ceramic Formulation Preparation
The precursors listed in Table I (without the actinides) were weighed and mixed in the amounts to form the desired precursor batch size. The batch was added to a polyethylene bottle filled approximately half full with 0.64 mm (0.25 inch) cylindrical calcium stabilized zirconia grinding media. Deionized water was added to just cover the batch and grinding media. The mixture was then ball milled for about 20 hours. The milled slurry was poured through a #40 sieve opening to catch the grinding media and allow the slurry to collect in a drying pan. The solution was then dried at 105°C so that no moisture was retained. The dried material was removed from the pan and calcined at 750°C for 1 hour in a shallow alumina tray. After cooling, the precursor batch was set aside for later mixing with PuO2 and UO2 to form the baseline composition or mixing with CeO2 as a surrogate for the actinides.
For the baseline formulation, PuO2 and UO2 powders were weighed out and added to the appropriate amount of precursor batch to coincide with the baseline composition (Table I). This formulation was referred to as Hf-Pu-U. For the surrogate baseline formulation, CeO2 as a surrogate for both PuO2 and UO2 on a one-to-one molar basis was added to the appropriate precursor batch amount. The surrogate formulation was referred to as Hf-Ce-Ce. Hf-Pu-U and Hf-Ce-Ce batches were added to a polyethylene bottle filled approximately half full with 1/4 inch cylindrical calcium stabilized zirconia grinding media. Deionized water was added to just cover the batch and grinding media. The batches were milled for approximately 8 hours. The slurry was screened to remove the grinding media and dried on a hot plate. The resulting cake was crushed and forced through a #40 sieve screen. The powder was then used for thermal analysis or for pressing into pellets and subjected to isothermal heat treatments. Pellets (approximately 1 g) were pressed using uniaxial compression at a pressing pressure of 13.8 MPA (2000 psi) in 12.7 mm (0.50 inch) dies. The diameter, thickness and mass of each pellet were measured after pressing.
Thermal Analysis
Differential thermal analysis (DTA) and thermogravimetric analysis (TGA) were run (in duplicate) on samples of the Hf-Ce-Ce batch to identify reaction temperatures. Typical scan rates of 10°C/min were used in all of the testing. The DTA scans were run to the nominal sintering temperature of 1350°C. Due to instrument limitations, the TGA scans were run only to 1200°C in these initial tests. The results were also evaluated using the first derivative to aid in the identification of reactions. For the Hf-Pu-U composition, TGA and differential scanning calorimetry (DSC) were used in an attempt to quantify reaction temperatures. DSC runs were only made to 700°C due to equipment limitations.
Toward the end of this study, a simultaneous DTA/TGA was available and samples of Hf-Ce-Ce baseline powder were run at 10°C/min to 1350°C in air, oxygen and helium atmospheres.
Isothermal Heat Treatments
Once potential reaction temperatures were determined, a series of isothermal heat treatments at temperatures slightly above those depicted in the thermal analysis scans were run on the material. In areas in the thermal analysis scans that were difficult to discern (perhaps due to multiple reactions occurring), several additional temperatures were selected for testing. Two pellets of both Hf-Ce-Ce and Hf-Pu-U compositions were heat-treated for two hours at the prescribed temperature and then quenched. Pellets were placed on reticulated yttria stabilized zirconia setters for the heat treatments to preclude interactions with the setter.
Sample Characterization
Following heat treatment, the diameter, thickness and mass for each pellet was measured. From these measurements, the geometric density was calculated for each pellet. X-ray diffraction scans were run on a sample from each of the prescribed heat-treatments to determine the phases present.
Results and Discussion
The phases present in the Hf-Pu-U isothermally heat-treated samples are shown in Table III. The results of the isothermal heat treatments for the Hf-Ce-Ce samples are shown in Table IV.
Upon heating, both compositions exhibited similar behavior with respect to the Ca(OH)2 decomposition, in-growth of CaCO3 and decomposition of CaCO3. This behavior in the Pu immobilization ceramic has been previously described [4].
Hf-Pu-U System
In the 600°C heat treatment in the Hf-Pu-U system, a calcium uranium oxide phase (CaUO4) was evident. This phase continued to increase in relative concentration with additional heat treatments until 950°C and then disappeared from the phase assemblage in the 1100°C heat treatment sample. Examination of the CaO-UO2 phase diagram (which indicated extensive solid solubility between CaO and UO2) [5] and subsequent heat treatments in the CaO-UO2 binary system confirmed the formation of this phase. Not surprisingly, UO2 was also found to oxidize upon heating in air to form U3O8. However, by 900°C no uranium oxide phase was detected in the samples. An additional low temperature uranium bearing phase, brannerite, also appeared to form at relatively low temperatures (750°C) and then be consumed in further development of the microstructure at 900°C. The appearance and disappearance of the brannerite phase was surprising since it was observed to reappear at higher temperatures (1200°C) and is always present (at about 10 vol %) in the Hf-Pu-U baseline ceramic upon sintering at 1350°C. It is quite possible that the composition of the "low temperature" and "high temperature" brannerite phases differ. Future microscopic and chemical analyses will be aimed at examining this hypothesis.


In the 950°C heat treatment, zirconolite was observed as well as the initiation of the anatase to rutile phase transformation. At 1025°C, no zirconolite was evident in the XRD patterns, however, pyrochlore formation was observed. The dimensional measurements of the samples (see below) indicated that densification in the ceramic coincides with the formation of the pyrochlore phase. At 1100°C, all anatase has converted to rutile and the only other oxides from the initial batch that were not completely reacted are HfO2 and PuO2. At 1200°C all the HfO2 had reacted and by 1250°C all the PuO2 had reacted.
Hf-Ce-Ce System
At 725°C, a perovskite phase formed in the Hf-Ce-Ce system. The relative concentration of a perovskite phase appeared to be relatively constant until it disappeared at 1100°C. A perovskite phase was observed to reappear at 1300°C and increase in relative concentration in the final sintered product at 1350°C. Similar to the behavior with brannerite in the Hf-Pu-U composition, the perovskite behavior is puzzling since it is always observed in the Hf-Ce-Ce composition in discernible concentrations. The potential for differing compositions of the "low temperature" and "high temperature" perovskite phases will also be examined in future microscopy studies. It is known that Ce readily changes oxidation states from Ce4+ to Ce3+ at elevated temperatures [6]. Thermal analysis studies (see below) confirmed this behavior in this system. The "high temperature" perovskite phase could very likely result from the reduction of Ce and the "ejection" of the Ce3+ from the pyrochlore phase.
At 900°C, the initiation of pyrochlore formation in the Hf-Ce-Ce ceramic was observed. The relative concentration of pyrochlore increased in subsequent heat treatments at 950°C and 1025°C. After the 1100°C heat treatment pyrochlore was the major crystalline phase in the assemblage. Dimensional measurements again pointed to the formation of pyrochlore coinciding with densification (see below).
Similar to the Hf-Pu-U system, the anatase to rutile transition was observed to initiate in the 950°C heat treatment and the conversion was completed following the 1100°C treatment. The HfO2 was also observed to all be reacted by 1200°C. Finally, CeO2 was found to behave very similarly to PuO2 in that complete dissolution of the CeO2 was also found to be completed by 1250°C.
Zirconolite was also observed to form in the Hf-Ce-Ce ceramic at 1200°C and remain in the ceramic at the final sintering temperature. Similar to the "high temperature" perovskite phase, the reduction of cerium is also very likely responsible for stabilizing the zirconolite phase.
Densification
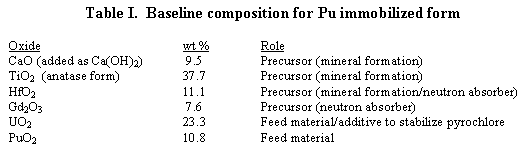
The densification behavior was examined by performing measurements on the samples before and after heat treatments. Figure 1 shows the fractional diameter vs. the heat treatment temperature. The fractional diameter is the diameter after heat treatment at the prescribed temperature divided by the original diameter.
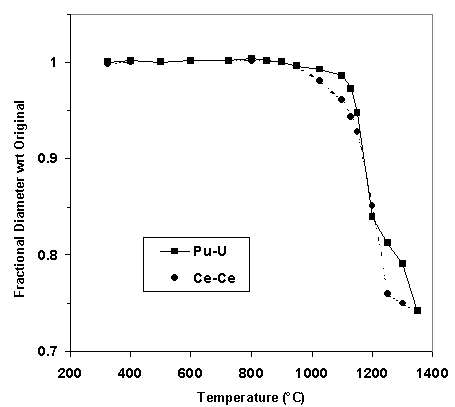
Figure 1. Densification behavior of Hf-Pu-U and Hf-Ce-Ce ceramics
expressed as
the fraction obtained from the diameter of the pellet divided
by the original diameter at the prescribed temperature.
When comparing the phase development results to the densification behavior it was apparent that major densification coincides with pyrochlore formation in both systems. Densification in the Hf-Ce-Ce began at a significantly lower temperature than in the Hf-Pu-U system (approximately 900°C vs. 1050°C for the Hf-Ce-Ce and Hf-Pu-U systems, respectively). In both systems, it was near these respective temperatures that pyrochlore formation was initiated.
It was also noted that the phases comprising the final assemblages were present by 1250°C in the Hf-Pu-U system and by 1300°C in the Hf-Ce-Ce systems yet significant densification occurred after these temperatures. This was especially evident in the Hf-Pu-U ceramic. Future microscopic analysis will be used to evaluate the microstucture development at these temperatures.
It was also interesting that the dimensional changes of the pellets in both systems following sintering at the baseline sintering temperature of 1350°C were identical. Although the end-points were the same, it was obvious from the shape of the curves that the densification paths were different for the two systems. It is possible that the formation of brannerite is retarding sintering in the Hf-Pu-U system. Future dilatometry studies and thermokinetic analysis are planned for these systems with the intention of modeling the sintering behavior in the two systems. This should provide insight into the sintering mechanisms occurring in the two systems.
Thermal Analysis
When it was determined that the reduction of cerium may be playing an important role in the phase development in the Hf-Ce-Ce composition, a few scoping experiments were performed in an attempt to verify the cerium reduction. Thermogravimetric scans were run on samples of the Hf-Ce-Ce composition in air, oxygen and helium atmospheres (Fig. 2).
The weight loss in the TGA scans at the higher temperatures pointed to the reduction of cerium. The fact that this behavior initiated at a lower temperature and the loss was exaggerated in the sample run in helium further confirmed the hypothesis. Additional TGA work with a coupled mass spectrometer is planned to quantify this behavior. Efforts are also underway to determine the cerium speciation (i.e. ratio of Ce3+ to Ce4+). The weight gain at lower temperatures in the sample heated in air was observed previously and is associated with in-growth of calcium carbonate [4].
Figure 2. Thermogravimetric analysis scans of Hf-Ce-Ce sample heated in different atmospheres.
Conclusions
Cerium oxide as a surrogate for the actinide oxides in the Pu immobilized form is adequate from many perspectives and is, thus, a suitable "stand-in" for process development studies. Although not discussed here, cerium oxide performed well as a physical surrogate for batching, powder handling and compaction. In both systems, the major phase formed was pyrochlore and the dissolution behavior of cerium oxide and plutonium oxide into the crystalline assemblage generally coincided. Finally, the overall shrinkage of the two systems under the baseline sintering conditions was essentially identical.
There were, however, significant differences between the behavior of cerium oxide and the actinide oxides that limits the use of cerium oxide as a true chemical surrogate. The response of calcium varied between the two systems. In the Hf-Pu-U system, calcium reacted at low temperatures to form a calcium uranium oxide phase whereas in the Hf-Ce-Ce system calcium reacted at a slightly higher temperature to form perovskite. The formation of pyrochlore and the onset of densification occurred at a lower temperature in the Hf-Ce-Ce composition. The most pronounced limitations appeared to be associated with the reduction of Ce at elevated temperatures. The reduction was undoubtedly responsible for the occurrence of zirconolite and perovskite in the final phase assemblage in the Hf-Ce-Ce formulation. The cerium reduction also appeared to have an effect on the densification behavior in the Hf-Ce-Ce ceramic and may be responsible for different sintering mechanisms occurring in these systems.
Acknowledgments
The information contained in this paper was developed during the course of work under Contract No. DE-AC09-96SR18500 with the U.S. Department of Energy.
References