
WSRC-RP-2000-00980
Discovery of New Plutonium Chemistry and Its Potential
Effect on LLW Disposal at SRS
D. I. Kaplan and E. L. Wilhite
Westinghouse Savannah River Company
Aiken, SC 29808
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
This report has been reproduced directly from the best available copy.
Available for sale to the public, in paper, from: U.S. Department of Commerce, National Technical Information Service, 5285 Port Royal Road, Springfield, VA 22161, phone: (800) 553-6847, fax: (703) 605-6900, email: orders@ntis.fedworld.gov online ordering: http://www.ntis.gov/support/ordering.htm
Available electronically at http://www.osti.gov/bridge/
Available for a processing fee to U.S. Department
of Energy and its contractors, in paper, from: U.S. Department of Energy, Office
of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831-0062,
phone: (865 ) 576-8401, fax: (865) 576-5728, email: reports@adonis.osti.gov
1. Summary
Recently published work on the chemistry of plutonium (IV) dioxide has shown that PuO2 is not the thermodynamically stable form as was previously thought. In humid environments or the presence of water, some of the plutonium is oxidized to Pu (VI) with the evolution of hydrogen and the formation of PuO2+x, where x can range up to 0.27 (i.e. about 27% of the Pu has been oxidized to the +6 oxidation state). The implication of this discovery is that a more mobile form of plutonium (i.e. Pu (VI)) than that assumed in the performance assessment (i.e. Pu (IV)) could be present in the humid waste disposal environment. If so, then the approved performance assessment might not conservatively bound the effects of plutonium waste disposal.
Based on SRS studies and literature data, this evaluation concludes that the discovery will not impact LLW disposal at SRS because most Pu (VI) that might form will likely be rapidly reduced to Pu (IV) before it could migrate far from the disposed waste and because the plutonium Kd used in the performance assessment is likely a conservative representation of the mix of plutonium oxidation states that could exist in SRS waste. Nonetheless, additional SRS studies are recommended, as a component of performance assessment maintenance, to demonstrate conclusively the fate of Pu (VI) in the SRS environment.
Keywords: Performance Assessment, Evaluation, Unreviewed Disposal Question
2. Introduction
One intent of DOE Order 435.1, U.S. DOE. (1999a), as expressed in the performance assessment/composite analysis guidance, U.S. DOE. (1999b), is to ensure that proposed or discovered changes in wasteforms, containers, radionuclide inventories, facility design, and operations are reviewed to ensure that the assumptions, results, and conclusions of the DOE approved performance assessment (PA), WSRC (2000), and composite analysis (CA), WSRC (1997), as well as any special analyses (SA) that might have been performed, remain valid (i.e. that the change or discovery is bounded by the PA and CA) and the change or discovery is within the bounds of the Disposal Authorization Statement, U.S. DOE. 1999c. The goal is to provide flexibility in day-to-day operation and to require those issues with a significant impact on the PA's conclusions, and therefore the projected compliance with performance objectives/measures, to be identified and brought to the proper level of attention. It should be noted that the term performance measure is used to describe site specific adaptations of the DOE Order 435.1 Performance Objectives and requirements (e.g. performance measures such as applying drinking water standards to the groundwater impacts assessment).
The intent of this document is to determine if the discovered change in plutonium chemistry (i.e. oxidation of PuO2 to form plutonium in the more mobile hexavalent form, Haschke et al. (2000)) is within the assumptions, parameters, and bases of the approved PA, WSRC (2000) and CA, WSRC (1997). If it is, then this document serves as the technical basis for continued disposal of LLW containing Pu. If not, then in order to continue Pu-bearing LLW disposal, the PA and CA would need to be updated as appropriate and DOE approval sought for the update (special analysis or revision of the PA or CA). Alternatively, compensatory measures (e.g. restricting disposal of Pu-bearing waste) could be taken.
3. Description of Discovery and Implication for LLW Disposal
Over the past few years, work on the chemistry of plutonium (IV) dioxide by J.M. Haschke and others has shown that PuO2 is not the thermodynamically stable form as had previously been understood, Haschke et al. (2000). Rather, in the presence of water, some of the plutonium dioxide surface is oxidized to Pu (VI) with the evolution of hydrogen and the formation of PuO2+x where x can range up to 0.27 (i.e. about 27% of the Pu has been oxidized to the +6 oxidation state). This reaction produces a more mobile form of plutonium (i.e. Pu (VI)) than that assumed in the performance assessment (i.e. Pu (IV)). The reaction is:
PuO2(s) + xH2O(ads.) ® PuO2+x(s) + xH2(g)
Because of the potential deleterious effects of this phenomenon on a variety of SRS programs (e.g. Pu immobilization, LLW disposal), a group of selected personnel representing plutonium science, plutonium programs and waste disposal and environmental restoration interests was formed to consider the implications of this discovery.
The group consensus, Meadors and Wilhite (2000), was that the work is technically credible. However, the chemical reaction reported by Haschke et al. to generate PuO2+x has a slow reaction rate. To produce significant quantities of Pu (VI) would require several years (i.e. 10 years to generate 5% Pu (VI) assuming a 20 m2/g surface area). Haschke's work was done using plutonium oxide with a higher surface area than that normally encountered at SRS.
The group agreed that the production of hydrogen by Haschke's mechanism is very small with respect to that produced by radiolysis. Radiolytic hydrogen production is considered in SRS processes (e.g. Pu storage, waste shipment to WIPP), thus, hydrogen production should not be a concern according to Haschke's mechanism.
With respect to the production of Pu (VI), the group agreed that hexavalent Pu would not present a concern for SRS programs (e.g. Pu storage, waste shipment to WIPP, Pu stabilization, Pu immobilization, MOX fuel) other than waste disposal and environmental restoration.
For waste disposal (i.e. LLW at SRS, WIPP, Yucca Mountain) and environmental restoration, the presence of Pu (VI) is a potential concern that must be addressed. Plutonium (VI) species are more soluble and more mobile in the environment than Pu (IV) species. Plutonium (IV) is the oxidation state that was generally assumed in assessments of disposal facility and remedial action performance.
4. Supporting Analysis
4.1 Influence of Oxidation State on Pu Geochemical Behavior
The objective of this section is to provide an overview of plutonium geochemistry and to also provide a review of what is known about plutonium geochemistry at SRS. Particular attention was directed at evaluating the influence of oxidation state on plutonium sorption to SRS sediments. The conclusion of this section provides an appraisal of the likely environmental fate of Pu (VI) at SRS.
4.2 Overview of Pu Geochemistry
4.2.1 Aqueous Chemistry
The single most important parameter controlling plutonium aqueous chemistry is its oxidation state. Plutonium exists in five oxidation states in aqueous solutions that include Pu (III), Pu (IV), Pu (V), Pu (VI), and Pu (VII). These states essentially always occur as the hydrated ions Pu +3, Pu+4, PuO2+, PuO2+2, and PuO4-1 (Cleveland 1979). It has been shown that Pu (III), Pu (IV), Pu (V) and Pu (VI) can coexist in unequal quantities in the same solution. With redox potential alone (i.e. without taking complexation reactions and other chemical reactions into consideration), it is difficult to predict stable oxidation states of plutonium ions present in a given water. Disproportionation (Disproportionation is a chemical reaction in which a single compound serves as both oxidizing and reducing agent and is thereby converted into a more oxidized and more reduced derivative.) also influences plutonium oxidation state. For example, Pu (V) ions can disproportionate to Pu (IV) and Pu (VI) (Choppin et al. 1997).
Another important chemical reaction controlling plutonium speciation is hydrolysis. Hydrolysis reactions lead to the formation of positively charged to neutral colloidal hydrolyzed molecules in a stepwise manner. For example,
Pu+4 + OH- ® PuOH+3 + OH- ® Pu (OH)2+2 etc.
leads finally to Pu(OH)4 which loses water to produce PuO2 (Cleveland 1979). The kinetics of the hydrolysis reaction leading to PuO2 are not known. Depolymerization of colloidal Pu (IV) is a very slow process, having a "depolymerization" half-life of 320 hr at 25 ° C in 5 M HNO3 (Cleveland 1979). Complexing agents such as fluoride or sulfate ions can accelerate the process. The tendency of plutonium to hydrolyze follows the effective charge of the ion in the order of (Kim, 1986):
Pu+4 > PuO22+ > Pu+3 > PuO2+.
Ions of plutonium in the (V) and (VI) states exist in the plutonyl form. The fact that the PuO22+ ion has an effective charge greater than the Pu3+ ion may be explained by a linear structure of the (O-Pu-O)2+ ion, in which the charge of the equatorial side becomes intensively exposed. Such an effect results in the effective charge of PuO2+ and PuO22+ corresponding to +2.3 ± 0.2 and +3.3 ± 0.1, respectively (Kim, 1986).
Plutonium can form complexes with most of the ions commonly encountered in sediment solutions. These anions include HCO3-/CO32-, Cl-, SO42-, PO43-, and F-. The propensity of various anions to form complexes with actinides of varying oxidation states is presented in Figure 1. The y-axis in the figure is an association constant (b 11); the greater its magnitude, the stronger the bond for the Pu complex. Generally the complexation of anions with Pu (IV) and Pu (VI) is greater than with Pu (III) and Pu (V). The overall tendency, across all oxidation states, to form complexes with these anions can be summarized as follows, from strongest to weakest complex:
CO32- > OH- > Humic Materials > F- > SO42- > PO43- > Cl-.
SRS groundwater is dominated in decreasing order of anion concentration by:
HCO3- > SO42- > Cl- = NO3-.
Not included in the above listing is humic materials, whose concentration varies greatly from <0.1 mg of carbon per liter in many subsurface environments to 50 mg of carbon per liter in wetland environments. In environments impacted by the presence of cement or grout, the dominant anions become heavily skewed towards OH- and HCO3-, which are strong Pu complexants. The implications of these complexes on plutonium sorption to sediments are described below.
Thus, disproportionation, complexation, and hydrolysis reactions combine to make plutonium chemistry exceptionally complicated. Because of this complexity, few generalizations commonly assigned to other elements are applicable to plutonium. An Eh (the oxidation-reduction potential, measured in volts) – pH equilibrium diagram of a simple system is depicted in Figure 2 in which 10-7 M Pu, water, and CO2-containing air are present. Conditions for natural aquatic solutions are within the inscribed window in the middle of the figure. It is important to keep in mind that this is a simplified representation of the likely plutonium geochemistry. Important reactions, such as humate material complexation, sulfate complexation, and disproportionation (especially in the lower pH region), are not represented in Figure 2. Whenever a complexation is involved (e.g. carbonate complexation) the oxidation potentials of Pu change considerably (Kim 1986). As the pH increases, the dominant plutonium species within the inscribed window progress from being free ions (Pu3+), to hydrolysis species [Pu(OH)3+ and PuOH2+], to carboxyl-hydroxyl [Pu(OH)3CO3- and Pu(CO3)33-] species in the high pH environment. The dominant oxidation state of plutonium in natural aquatic condition is primarily Pu (IV), and to a lesser extent, Pu (III) and Pu (V). Importantly, Pu (VI) is not stable except in extremely oxidized and acidic environments, typically not found in the natural environment.
4.2.2 Sediment Sorption
Sediment redox potential (which can be represented as Eh) has a profound influence on the tendency of plutonium to sorb to sediments. A number of investigators have examined sorption of plutonium on minerals, soils and other geological substrates. The importance of plutonium redox status on sorption was demonstrated by Bondietti et al. (1975) who reported about 2 orders-of-magnitude difference in Kd values with montmorillonite between Pu (VI) (250 mL/g) and Pu (IV) (21,000 mL/g).
Once in the subsurface environment, there are many naturally occurring materials that can change the oxidation state of plutonium. Natural dissolved organic matter (DOC) (e.g. fulvic and humic acids) can reduce plutonium from the +6 state to the +4 state (Bondietti et al. 1975; Nelson et al. 1987; Choppin and Morse 1987). Similarly, naturally occurring Fe-oxides have been reported to reduce plutonium from the +5/+6 state to the +4 oxidation state in what is possibly an irreversible reaction (Sanchez et al. 1985; Keeney-Kennicutt and Morse 1985). Conversely, Mn-oxides have been shown to potentially oxidize plutonium from the +4/+5 state to the +6 state (Duff et al. 1999).
Krupka et al. (1999) concluded that redox status, pH, and carbonate concentration were the aqueous parameters that had the greatest impact on plutonium sorption. Complexation of plutonium by various ligands can significantly influence its sorption behavior (Benson 1960, Relyea and Brown 1978). Increasing concentrations of EDTA, acetate, and oxalate ligands resulted in decreasing sorption of plutonium onto sediments (Benson 1960). Increasing concentrations of carbonate ligands have an especially strong impact on depressing plutonium sorption to mineral surfaces (Sanchez et al. 1985).
Sorption experiments conducted by Billon (1982) indicated Kd values for Pu (IV) ranging from 32,000 to 320,000 mL/g (depending on pH) for bentonite or attapulgite. Additional experiments conducted with Pu (VI) species on bentonite substrate resulted in Kd values ranging from about 100 to 63,100 mL/g when pH was varied from 3.1 to 7.52. Sanchez et al. (1985) compared the sorption of Pu (IV) and Pu (V) onto goethite (i.e. FeOOH2). They found that the free ion species (Pu4+ and PuO2+) and the various complexed-plutonium species sorbed directly to the ionized hydroxyl sites of the goethite. The sorption isotherm obtained at a higher (10-10 M) compared to a lower (10-11 M) concentration of total soluble Pu (IV) and Pu (V) showed that the adsorption edges (i.e. pH value at which 50% sorption occurs) increased towards a higher pH value. This indicates that the Kd value is highly dependent on the amount of Pu (IV) or Pu (V) in solution, contrary to the Kd construct that requires independence from the solute concentration. This data also showed that the sorption curve for Pu (V) shifted ~2 pH units higher as compared to the sorption curve observed for Pu (IV), indicating that Pu (V) had lower sorbing affinity than the Pu (IV).
Sorption of plutonium in +4, +5, and +6 oxidation states on a Hanford Site shallow sediment was studied by Barney (1992) to elucidate any differences in rate and amount of sorption of plutonium in different oxidation states. The Kd values range from:
2100 to 11,600 mL/g for Pu (IV),
2100 to 4,600 mL/g for Pu (V), and
1000 to 4,600 mLg for Pu (VI).
4.2.3 Solubility
In high pH environments such as several of the disposal units analyzed in the PA (i.e., the low activity waste (LAW) vaults, the intermediate level (IL) vaults, the intimately-mixed cement-stabilized waste (ashcrete) trenches, and the cement-stabilized encapsulated waste (components in grout) trenches), the solubility of plutonium will be greatly reduced. Under the conditions in these waste disposal units, Pu (IV) will be the predominant species. Plutonium (VI) will not exist under these circumstances (Nelson, et al 1989). Thus, the new plutonium chemistry will not impact the PA assumptions regarding Pu solubility.
4.3 Review of Pu Geochemical Studies Conducted at SRS
There have been six studies conducted on plutonium geochemistry at the SRS: Prout (1958), Relyea and Brown (1978), Hawkins (1985), Gibbs et al. (2000), Wilhite (1978), and Kaplan et al. (1994). Additionally, Hoeffner (1985) provided a brief review of Pu geochemistry at the SRS. The first four studies evaluated the magnitude of plutonium sorption to SRS sediments, whereas the last two studies provided some insight as to the oxidation state of plutonium in an SRS aquifer.
Prout (1958) studied sorption of plutonium in +3, +4, and +6 oxidation states to an SRS soil as a function of pH (Figure 3). The calculated Kd values ranged from:
<10 to >10,000 mL/g for Pu (III),
~100 to ~10,000 mL/g for Pu (IV), and
<10 to ~3,000 mL/g for Pu (VI).
Maximum Kd values were observed between pH values of 6.5 and 8.5. Because the initial concentration of plutonium used in these experiments was about 10-6 M, it is very likely that precipitation occurred during these studies. Nelson et al. (1987) showed that plutonium precipitation occurred if the solution concentration exceeded 10-7 M. This significantly compromises the data for its use for describing adsorption or cation exchange processes, the dominant processes assumed to be described by Kd values used in contaminant transport calculations.
Relyea and Brown (1978) used a sand-textured Fuquay sediment from SRS to evaluate Pu (IV) sorption. The initial Pu (IV) was 5 x 10-8 M, which suggests that homogeneous precipitation likely did not occur during these experiments. The Kd value was 316 mL/g. When 10-3 M of EDTA was added to the system, the measured Kd value decreased to 120 mL/g. This significant reduction in plutonium sorption was attributed to the limited affinity of Pu-EDTA complex to sorb onto the soil mineral surfaces. Increasing the EDTA concentration by an order of magnitude resulted in further reduction in Kd values by two orders-of-magnitude: Kd was 0.12 mL/g. Kd values calculated from desorption experiments showed that the Kd values were 1 or 2 orders of magnitude higher than the values calculated from the adsorption experiments.
Hawkins (1985) placed filters containing Pu (VI) in the subsurface of mini-lysimeters filled with SRS sediments. The Pu (VI) was exposed to natural weathering conditions for two years, during which time 79-L of rainwater percolated through the mini-lysimeters. The mini-lysimeters were then sampled as a function of depth below the Pu (VI) source. Most of the Pu was found within the 1-inch soil section containing the spiked Pu (VI). The Kd value estimated from the sample results varied between 8 and 35 mL/g. Less retention (lower Kd values) was calculated with increasing distance from the source. This suggested to the Hawkins (1985) that the Pu had converted to more than one oxidation state, whereby the lower Kd values were due to the presence of Pu (VI) and the higher Kd values were due to the presence of Pu (IV) or Pu (III). However, the entire range of Kd values they calculated from this experiment is quite low, suggesting that in fact, none of the Pu had been reduced to the +4 or +3 state, which tend to have Kd values >1000 mL/g. Hawkins used a rather indirect method of calculating Kd values from this data. Instead of using the usual method of calculating Kd values whereby all the Pu soil data is used to generate one Kd value (actually a retardation factor), he calculated separate Kd values for each depth. Pu Kd values need to be calculated from this data using the traditional approach.
Conversely, Gibbs et al. (2000) conducted a series of Pu (V) soil column experiments. Based on the sorptive behavior of the plutonium, they concluded that the plutonium had been converted to Pu (IV) during the course of the experiments.
Wilhite (1978) conducted a detailed study on the form of plutonium in the groundwater and soil of the burial ground. He determined that about half of the plutonium in the groundwater could be filtered (<120 nm), suggesting that it was in a colloidal state. This is approximately the same proportion of plutonium Kaplan et al. (1994) reported could be filtered from F-Area groundwater. Wilhite (1978) also reported that 90% of the plutonium was cationic, 4% was anionic, and the remainder was presumably neutrally charged. Especially relevant to this discussion is his finding that 30% of the plutonium existed as Pu (III), 25% as Pu (IV), 43% as Pu (VI), and <2% as an organic-Pu complex. He also measured the following Kd values:
The three in-situ Kd values were two to three orders-of-magnitude greater than the batch Kd value. The large in-situ values likely reflect the fact that the Pu had time to "age" with the sediment, thereby having time to diffuse into the solid structures (especially of Fe or Al coatings). It may also be the result of the Pu having sufficient time to convert from Pu (VI) to Pu (IV). The large Kd values are consistent with the observation that most of the Pu in the groundwater existed in oxidation states that strongly sorb, i.e., the +3 and +4 states. Of these values, the larger in-situ Kd values are likely more representative of the conditions expected in a performance assessment scenario.
Kaplan et al. (1994) concluded that the plutonium emanating from the F-Area Seepage Basin existed primarily in the +4 oxidation state. They based this conclusion on the observation that the plutonium in groundwater samples partitioned to mobile colloids in a manner similar to thorium (a +4 element) and not like curium (a +3 element) or uranium (a +6 element in this system). By analogy, this data provided indirect evidence that the plutonium existed primarily in the +4 state.
Figure 4 presents the dominant oxidation states of plutonium as a function of pH and the inscribed area in the figure represents the pH and Eh levels in most soils, similar to that presented in Figure 2. As was concluded from Figure 2, plutonium exists primarily in the +3 oxidation state in pH environments <4.5, in the +3, +4 and +5 state between pH 4.5 to 7, and in the +4 state between pH 7 to 8.5 environments. In environments with a pH >8.5, such as in a cement-impacted environment, the dominant plutonium oxidation state will be +4. Also included in Figure 4 are six Eh-pH data points measured in groundwater samples collected from H-Area (Stone et al. 1984). These data suggests that plutonium at equilibrium with SRS groundwater would exist primarily as Pu (IV) and Pu (III).
4.4 Interpretation
At issue in this report, is whether the potential formation of Pu (VI) in disposed waste will significantly affect the manner that plutonium geochemistry should be modeled in performance assessments and risk calculations. In the past, plutonium was modeled with the implicit assumption that it existed in the +4 oxidation state. Plutonium in the +4 oxidation state sorbs appreciably stronger to sediments than in the +6 state. Based on the findings of Wilhite (1978), which is consistent with the findings of several other researchers at other locations, it is likely that Pu exists in our groundwater in several oxidation states and that if Pu (VI) is introduced into our subsurface, it will in part be reduced to Pu (III) and Pu (IV). Lines of evidence in support of these contentions follow.
Some evidence exists that contradicts the notion that Pu (VI) will be totally reduced in the SRS environment. The fact that Hawkins (1985) observed high mobility of Pu in mini-lysimeters after it had been introduced as Pu (VI) is troubling, in that it suggests that the Pu (VI) was not converted to the less mobile Pu (IV). Additionally, Wilhite (1978) measured Pu in the burial grounds in the +3, +4 and +6 oxidation states. The oxidation state of the Pu waste when it was first introduced into the ground is not known. Another potential problem with assuming that Pu (VI) will be reduced is the abundance of Mn-oxides in SRS sediments. It is not known whether these minerals can oxidize Pu (IV) to Pu (VI), as Duff et al. (1999) demonstrated on Yucca Mountain tuff material.
Based on the discussion above, it appears likely that Pu (VI), if introduced into the SRS subsurface environment, would be entirely or partially reduced to Pu (IV) or Pu (III). The most compelling line of evidence is that SRS sediments are generally coated with Fe-oxides that can reduce plutonium, and that Pu (IV) was identified in an SRS groundwater by both Kaplan et al. (1994) and Wilhite (1978). Additional research is required to substantiate this interpretation.
Excluding Kd data that are either compromised due to experimental purposes or are not relevant to a performance assessment scenario (Prout entire data set and Wilhite’s batch sorption data), the following Kd values have been measured in SRS sediments:
Unfortunately, this data has an enormous range. The least reliable data in this list is that generated by Hawkins (1985) due to the indirect manner in which the Kd values were calculated. The most reliable data are the in-situ values generated by Wilhite (1978) due to the fact that these values capture the "aging" and the "desorption" processes, which are known to occur in nature. Given the above observations:
The PA assumed the Kd of Pu on SRS soil to be 100 mL/g. As shown above, this value is greater than the Pu (VI) conservative estimate, but less than the Pu (IV) or the likely Pu (III, IV, VI) conservative Kd estimates. Given the above considerations, and the likelihood that not all the Pu in our system exists in the +6 oxidation state, the Pu Kd used in the PA is likely a conservative value for Pu behavior. An alternative approach to modeling Pu mobility would be to break up the total Pu pool into fractions representing the various oxidation states. At this time, however, a definitive speciation of plutonium in the SRS subsurface environment has not been determined. Until such a determination is available, sensitivity/uncertainty analysis could be used to test the impact of varying proportions of the oxidation states.
5. Evaluation
6. Conclusion
Although the discovery reveals new aspects of plutonium chemistry that were not considered when the PA was prepared, this evaluation has shown that the new chemistry is not likely to affect the PA analyses and conclusions. The evaluation has shown that the new plutonium chemistry will not alter the solubility control employed in the PA for the cement vault waste disposal units (i.e., LAWV and ILV) or the cementitious wasteforms (i.e. ashcrete and components in grout). The evaluation concludes that a conservative plutonium Kd value for the mixture of oxidation states that are likely to exist in the SRS subsurface environment is 300 mL/g, which is greater than the 100 mL/g employed in the PA. Thus, no additional analyses or compensatory measures are needed to support continued disposal of LLW containing Pu, in any of the E-Area disposal units (i.e. vaults, trenches or NR pad).
Nonetheless, additional SRS laboratory studies are recommended, as a component of PA maintenance, to demonstrate conclusively the fate of Pu (VI) in the SRS environment. Additionally, the PA uncertainty program should examine the effect Pu speciation has on plutonium inventory limits.
7. References
(http://www.epa.gov/radiation/technology/partition.htm) (1999).

Figure 1. Comparison of complexation association constants (log B11) of EDTA, carbonate, hydroxide, humate, citrate, fluoride, sulfate and chloride with different actinide (An) oxidation states (Kim 1986).
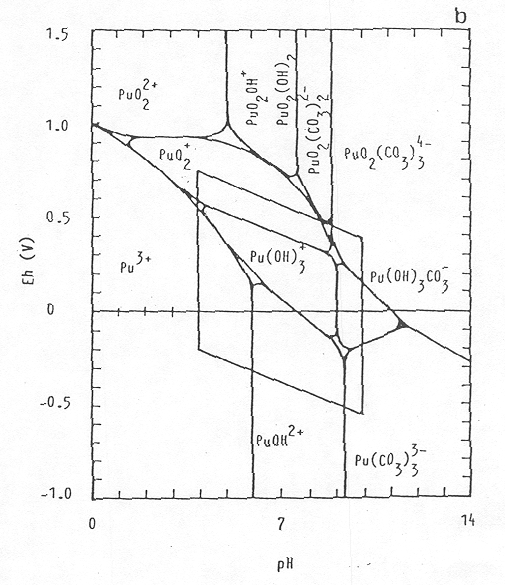
Figure 2. Eh - pH equilibrium diagram
of plutonium at 25 ° C (Kim 1986). The
inscribed window in the
center of the figure represents the pH and Eh levels in most soils and
was determined from a survey
involving 6,200 non-SRS data pairs (Baas-Becking et al. 1960).

Figure 3. Plutonium sorption as a function of pH and oxidation state on an SRS sediment (Prout 1958).
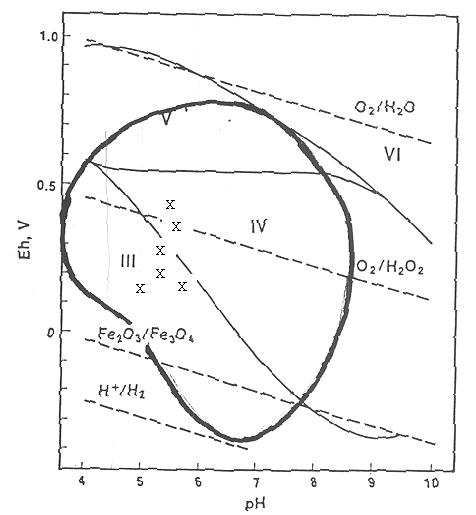
Figure 4. Eh-pH equilibrium diagram of the dominant oxidation states of plutonium (Morse and Choppin 1991). The inscribed area represents the pH and Eh levels in most soils and was determined from a survey involving 6,200 non-SRS data pairs (Baas-Becking et al. 1960). The six Eh-pH data points represent measurements taken from H-Area subsurface water (Stone et al. 1985).
